| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
Carnitine palmitoyltransferase I (CPT-I)
|
|---|---|
| 体外研究 (In Vitro) |
在 H9c2 细胞中,依托莫昔尔促进脂肪酸和甘油前体通过独特的代谢通道转化为心磷脂 [2]。尽管依托莫昔尔减少了[1-14C]棕榈酸或[1-14C]油酸掺入心磷脂,但它对参与心磷脂生物合成和重塑的酶的活性没有影响[2]。依托莫昔尔可增加心磷脂与[1,3-3H]甘油的结合。该过程涉及甘油激酶活性增加 33%,从而导致通过心磷脂生物合成从头途径的甘油通量增加 [2]。
当依托莫克不可逆地结合到 CPT-1 的催化位点时,它会抑制 CPT-1 的活性,同时增加移植氧化酶。 Etomoxir 被创建为线粒体外膜定位的线粒体肉毒碱支架扩增酶-1 (CPT-1) 的探针。 Etomoxir 通过充当氧化体增殖剂来刺激心肌中的 DNA 合成和心肌发育。因此,etomoxan 被视为除 CPT1 之外的 PPARα 激动剂。依托莫克西已被提议作为激活心脏突变的靶标。它是环氧乙烷激酶肉毒碱模板转移酶 I 家族的成员。肉毒碱模板转移酶 I 活性在依托莫克西治疗激活后不可逆地转录。因此,线粒体和β-氧化的碱基输入减少,导致细胞质积累和氧化增加。 Etomoxir 的长时间延迟(24 小时)对酶的表达甚至有明显的影响。 |
| 体内研究 (In Vivo) |
Etomoxir 可抑制 BMSC 分化的成骨细胞的减少,并显着抑制高脂肪 (HF) 和 db/db 饮食喂养的小鼠骨矿物质密度 (BMD) 和破骨强度的下降[3]。在喂食 HF 和 db/db 的小鼠中,依托泊昔抑制成骨细胞和小鼠线粒体 ROS 生成的增加[3]。依托莫克舍引起的体内肉碱棕榈酰转移酶-I (CPT-I) 部分抑制不会改变心脏长链脂肪酸的摄取和氧化速率[4]。
本研究评估了2型糖尿病患者游离脂肪酸(FFA)、ROS产生、线粒体功能障碍与骨密度(BMD)之间的关联,并探讨了其分子机制。db/db和高脂肪(HF)喂养的小鼠接受CPT1、MitoQ抑制剂Etomoxir和P53抑制剂PFT-α的治疗。评估骨代谢因素,分离BMSCs并诱导成骨分化。2型糖尿病患者的FFA、脂质过氧化和mtDNA拷贝数与BMD相关Etomoxir、MitoQ和PFT-α显著抑制了db/db和HF喂养的小鼠BMD和骨断裂强度的降低,并抑制了BMSCs分化成骨细胞的减少。Etomoxir和MitoQ,而不是PFT-α,抑制了db/db和HF喂养的小鼠和成骨细胞线粒体ROS产生的增加。此外,Etomoxir、MitoQ和PFT-α显著抑制了成骨细胞的线粒体功能障碍。此外,db/db和HF喂养的小鼠成骨细胞中的线粒体凋亡被激活,而Etomoxir、MitoQ和PFT-α则抑制了线粒体凋亡。此外,P53的线粒体积累募集Bax并引发凋亡事件的分子事件。这些结果表明,脂肪酸氧化导致ROS产生,激活P53/Bax介导的线粒体凋亡,导致T2DM成骨分化和骨丢失减少。[3] 尽管CPT-I(肉碱棕榈酰基转移酶-I)通常被认为是线粒体β氧化的主要速率控制位点,但CPT-I是否在心脏的整体LCFA(长链脂肪酸)通量中起限速作用尚不完全清楚。心脏中调节LCFA通量的另一个重要部位是CD36和FABPpm(质膜脂肪酸结合蛋白)促进的跨肌膜LCFA转运。因此,我们探讨了在LCFA-CoA线粒体进入水平上对LCFA通量的慢性药理学阻断在多大程度上会影响肌膜LCFA摄取。每天给大鼠注射生理盐水或依托莫西(一种特定的CPT-I抑制剂),持续8天,剂量为20mg/kg体重。etomoxir治疗的大鼠心脏CPT-I活性降低了44%。与对照组相比,依托莫西治疗的大鼠心脏中CD36和FABPpm的肌浆含量以及LCFA转运能力没有改变。此外,无论是在基础代谢需求下还是在急性诱导的最大代谢需求下,依托莫西治疗大鼠的LCFA摄取和氧化率以及心肌细胞对葡萄糖的摄取率与对照组大鼠没有差异。最后,依托莫西治疗大鼠的心脏没有显示出三酰甘油的积累。因此,CPT-I似乎不是心脏总LCFA通量的主要心率控制位点。肌膜LCFA进入而非线粒体LCFA-CoA进入可能是心脏代谢疾病中LCFA通量正常化的有前景的靶点[4]。 |
| 酶活实验 |
shRNA-GFP慢病毒质粒的构建与感染[1]
使用标准分子生物学技术,用GFP cDNA代替5个针对CPT1A的shRNA慢病毒质粒中的嘌呤霉素抗性基因。简言之,从慢病毒PELPS-GFP质粒中将1.4kb的GFP cDNA插入物亚克隆到pLKO.1 shRNA质粒中的Kpn和BamH位点。通过免疫印迹分析测定每个慢病毒质粒的效力。按照描述进行慢病毒感染36。将表达针对CPT1A的shRNA的淋巴细胞与相应的对照质粒进行比较,我们也将其改造为表达GFP而不是嘌呤霉素抗性。对照质粒编码来自人β-肌动蛋白基因的扰乱shRNA序列。实验中慢病毒感染的效率在60-90%之间。在细胞增殖评估中,在对GFP+细胞进行门控后,使用基于珠的计数方法进行计数。争夺GFP和shRNA-CPT1A-GFP病毒上清液的滴度分别为9.45e6和7.34e6 TU/ml。[1] 免疫印迹[1] 在shRNA慢病毒感染后5天评估CPT1A蛋白表达。在含有蛋白酶和磷酸酶抑制剂的RIPA-2(50mM Tris-HCl,pH8.0,150mM NaCl,1%NP-40,0.5%脱氧胆酸,0.1%SDS)中裂解细胞。通过SDS-PAGE分离等量的裂解物,并通过电泳将其转移到PVDF膜上。将膜在含0.1%吐温-20(TBS-T)的含5%脱脂乳的TBS(20mM Tris,135mM NaCl)中孵育1小时。封闭后,用含0.5%脱脂乳的含1:500稀释液的抗CPT1A探测膜。在TBS-T中洗涤一系列后,将膜与含1:10000稀释液的HRP缀合的山羊抗兔IgG孵育。使用West Femto SuperSignal化学发光试剂检测抗体结合。用1:2000稀释的小鼠抗β-肌动蛋白单克隆抗体,然后用1:5000稀释的HRP偶联的绵羊抗小鼠IgG测定相对蛋白质负荷。 |
| 细胞实验 |
细胞活力测定[2]
细胞类型: 大鼠心脏 H9c2 成肌细胞 测试浓度: 1-80 μM 孵育时间: 2小时 实验结果:减少了H9c2心肌成肌细胞中[1-14C]脂肪酸掺入CL和PtdGro,但不影响放射性的总掺入进入这些细胞。 |
| 动物实验 |
Animal/Disease Models: 80 male C57BLKS/J lar-Leprdb/db mice[3]
Doses: 1 mg/kg Route of Administration: Intraperitoneally injected; twice every week Experimental Results: Serum alkaline phosphatase was increased in db/db mice, which event was Dramatically suppressed by Etomoxir. Serum level of osteocalcin, a marker of bone formation, was decreased in db/db mice and Etomoxir markedly inhibited the reduction of osteocalcin. Serum tartrate-resistant acid phosphatase was elevated in db/db mice which phenomenon was Dramatically suppressed by Etomoxir. Animal/Disease Models: Rats[4] Doses: 20 mg/kg Route of Administration: Injected daily; for 8 days Experimental Results: Etomoxir-treated rats displayed a 44% decreased cardiac CPT-I activity. Male Lewis rats, weighing 150–200 g, were used in the present study. Animals were kept on a 12 h:12 h light/dark cycle and fed a Purina Chow diet and water ad libitum. The rats were divided into two groups: (1) control and (2) etomoxir. Etomoxir (20 mg/kg of body weight) was dissolved in 0.9% (w/v) NaCl and administered intraperitoneally for 8 days. Control rats received saline. The last injection was given 24 h before the experiment. Ethical approval for all experimental procedures was obtained from the Experimental Animal Committee of the Maastricht University, and the study conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Animals were anaesthetized with an intraperitoneal injection of a nembutal and heparin (3:1) mixture. Subsequently, the heart was removed for LCFA uptake studies and for analyses of transporter protein contents. [4] 80 male C57BLKS/J lar-Leprdb/db mice and 20 wild type littermates (8 week) were obtained from Model Animal Research Centre, Nanjing University, China. Mice were housed in cages in a limited access room, under temperature (23 ± 2 °C) and humidity (55 ± 5%) condition with a standard light (12 h light/dark) cycle and fed a regular diet. db/db mice were randomly divided into four groups: db/db group, Etomoxir group, MitoQ group, and PFT-α group. In the Etomoxir group, mice were intraperitoneally injected with 1 mg/kg Etomoxir twice every week. In the MitoQ group, 50 μmol/L MitoQ was given to the mice in water. Water bottles, containing either MitoQ, were covered with aluminum foil, and all bottles were refilled every 3 days. In the PFT-α group, mice were intraperitoneally injected with 1 mg/kg PFT-α twice every week. WT mice were administrated with vehicle instead. The experimental period is 8 weeks. At the end, peripheral blood samples and bone marrow cells were harvested for the assays. 100 C57BL/6 mice obtained from Experimental Animal Centre of Fourth Military Medical University. The mice were randomly divided into five groups: Control group, HF diet group, Etomoxir group, MitoQ group, and PFT-α group. Mice in HF diet, Etomoxir, MitoQ, and PFT-α groups were given high fat diet for 20 weeks and mice in Etomoxir, MitoQ, and PFT-α groups were administrated with Etomoxir, MitoQ, and PFT-α in the last 10 weeks. The administration of Etomoxir, MitoQ, and PFT-α were identical to the treatment in db/db mice. Control mice were administrated with vehicle instead [3]. |
| 参考文献 |
|
| 其他信息 |
Etomoxir (ETO) is a widely used small-molecule inhibitor of fatty acid oxidation (FAO) through its irreversible inhibitory effects on the carnitine palmitoyl-transferase 1a (CPT1a). We used this compound to evaluate the role of fatty acid oxidation in rapidly proliferating T cells following costimulation through the CD28 receptor. We show that ETO has a moderate effect on T cell proliferation with no observable effect on memory differentiation, but a marked effect on oxidative metabolism. We show that this oxidative metabolism is primarily dependent upon glutamine rather than FAO. Using an shRNA approach to reduce CPT1a in T cells, we further demonstrate that the inhibition of oxidative metabolism in T cells by ETO is independent of its effects on FAO at concentrations exceeding 5 μM. Concentrations of ETO above 5 μM induce acute production of ROS with associated evidence of severe oxidative stress in proliferating T cells. In aggregate, these data indicate that ETO lacks specificity for CTP1a above 5 μM, and caution should be used when employing this compound for studies in cells due to its non-specific effects on oxidative metabolism and cellular redox[1].
We examined the effect of etomoxir treatment on de novo cardiolipin (CL) biosynthesis in H9c2 cardiac myoblast cells. Etomoxir treatment did not affect the activities of the CL biosynthetic and remodeling enzymes but caused a reduction in [1-14C]palmitic acid or [1-14C]oleic acid incorporation into CL. The mechanism was a decrease in fatty acid flux through the de novo pathway of CL biosynthesis via a redirection of lipid synthesis toward 1,2-diacyl-sn-glycerol utilizing reactions mediated by a 35% increase (P < 0.05) in membrane phosphatidate phosphohydrolase activity. In contrast, etomoxir treatment increased [1,3-3H]glycerol incorporation into CL. The mechanism was a 33% increase (P < 0.05) in glycerol kinase activity, which produced an increased glycerol flux through the de novo pathway of CL biosynthesis. Etomoxir treatment inhibited 1,2-diacyl-sn-glycerol acyltransferase activity by 81% (P < 0.05), thereby channeling both glycerol and fatty acid away from 1,2,3-triacyl-sn-glycerol utilization toward phosphatidylcholine and phosphatidylethanolamine biosynthesis. In contrast, etomoxir inhibited myo-[3H]inositol incorporation into phosphatidylinositol and the mechanism was an inhibition in inositol uptake. Etomoxir did not affect [3H]serine uptake but resulted in an increased formation of phosphatidylethanolamine derived from phosphatidylserine. The results indicate that etomoxir treatment has diverse effects on de novo glycerolipid biosynthesis from various metabolic precursors. In addition, etomoxir mediates a distinct and differential metabolic channeling of glycerol and fatty acid precursors into CL. [2] This study evaluated the association between free fatty acid (FFA), ROS generation, mitochondrial dysfunction and bone mineral density (BMD) in type 2 diabetic patients and investigated the molecular mechanism. db/db and high fat (HF)-fed mice were treated by Etomoxir, an inhibitor of CPT1, MitoQ, and PFT-α, an inhibitor of P53. Bone metabolic factors were assessed and BMSCs were isolated and induced to osteogenic differentiation. FFA, lipid peroxidation and mtDNA copy number were correlated with BMD in T2DM patients. Etomoxir, MitoQ and PFT-α significantly inhibited the decrease of BMD and bone breaking strength in db/db and HF-fed mice and suppressed the reduction of BMSCs-differentiated osteoblasts. Etomoxir and MitoQ, but not PFT-α, inhibited the increase of mitochondrial ROS generation in db/db and HF-fed mice and osteoblasts. In addition, Etomoxir, MitoQ and PFT-α significantly inhibited mitochondrial dysfunction in osteoblasts. Moreover, mitochondrial apoptosis was activated in osteoblasts derived from db/db and HF-fed mice, which was inhibited by Etomoxir, MitoQ and PFT-α. Furthermore, mitochondrial accumulation of P53 recruited Bax and initiated molecular events of apoptotic events. These results demonstrated that fatty acid oxidation resulted in ROS generation, activating P53/Bax-mediated mitochondrial apoptosis, leading to reduction of osteogenic differentiation and bone loss in T2DM. [3] |
| 分子式 |
C15H18CLO4.NA
|
|---|---|
| 分子量 |
320.74
|
| 精确质量 |
320.079
|
| 元素分析 |
C, 56.17; H, 5.66; Cl, 11.05; Na, 7.17; O, 19.95
|
| CAS号 |
828934-41-4
|
| 相关CAS号 |
Etomoxir;124083-20-1; 828934-41-4 (sodium); 82258-36-4 (racemate)
|
| PubChem CID |
57345784
|
| 外观&性状 |
Typically exists as white to off-white solids at room temperature
|
| LogP |
2.188
|
| tPSA |
61.89
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
21
|
| 分子复杂度/Complexity |
321
|
| 定义原子立体中心数目 |
1
|
| SMILES |
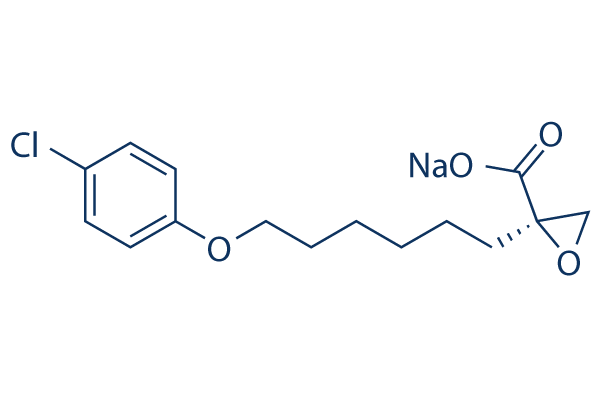
C([C@]1(OC1)C(=O)O)CCCCCOC1C=CC(Cl)=CC=1.[Na]
|
| InChi Key |
RPACBEVZENYWOL-XFULWGLBSA-M
|
| InChi Code |
InChI=1S/C15H19ClO4.Na/c16-12-5-7-13(8-6-12)19-10-4-2-1-3-9-15(11-20-15)14(17)18;/h5-8H,1-4,9-11H2,(H,17,18);/q;+1/p-1/t15-;/m1./s1
|
| 化学名 |
(2R)-2-[6-(4-chlorophenoxy)hexyl]-2-oxiranecarboxylic acid monosodium salt
|
| 别名 |
B 80754; B 807-54; B80754; B807-54; ETOMOXIR; 124083-20-1; R-(+)-Etomoxir; Etomoxir [INN]; UNII-MSB3DD2XP6; MSB3DD2XP6; ethyl (2R)-2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate; (R)-(+)-Etomoxir; B-807-54; B-80754
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: (1). 本产品在运输和储存过程中需避光。 (2). 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.5 mg/mL (7.79 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (7.79 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (7.79 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 3.33 mg/mL (10.38 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1178 mL | 15.5890 mL | 31.1779 mL | |
| 5 mM | 0.6236 mL | 3.1178 mL | 6.2356 mL | |
| 10 mM | 0.3118 mL | 1.5589 mL | 3.1178 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03144128 | COMPLETED | Dietary Supplement: Vitamin D Dietary Supplement: Placebo |
Cancer Cachexia Vitamin D Deficiency |
David Travis Thomas | 2018-05-23 | Not Applicable |
 马来酸哌克昔林
马来酸哌克昔林
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
