
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
Syk (IC50 = 7.7 nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
entopletinib (GS-9973) 在体外证明对 Caco-2 单层细胞具有良好的双向渗透性。 Entospletinib (GS-9973) 是一种基于细胞的药物,对 Syk 显示出良好的选择性,并有效抑制单核细胞中免疫复合物刺激的细胞因子产生以及 BCR 介导的 B 细胞活化和增殖 [1]。 Entospletinib (GS-9973) 和 Idelalisib 共同抑制 CLL 细胞活力并进一步阻碍趋化因子信号传导 [2]。
|
||
| 体内研究 (In Vivo) |
在大鼠和狗中,entespletinib (GS-9973)(1 mg/kg,口服)具有中等到高的生物利用度。在胶原诱导的关节炎大鼠模型中,entospletinib (GS-9973)(1-10 mg/kg,口服)可显着减少踝关节炎症。此外,Entospletinib (GS-9973) 在多项组织学测量中显示出疾病缓解活性,ED50 范围为 1.2 至 3.9 mg/kg[1]。这些测量包括抑制血管翳形成、软骨损伤、骨吸收和骨膜骨形成。
|
||
| 酶活实验 |
激酶测定[1]
以Lance为基础的测定格式测量全长杆状病毒表达的Syk激酶活性,最终体积为25μL,含有25 mM Tris-HCl,pH 7.5,5 mMβ-甘油磷酸,2 mM DTT,0.1 mM Na3VO4,10 mM MgCl2,0.5μM Promega PTK生物素化肽底物1,0.01%酪蛋白,0.01%Triton X-100,0.25%甘油和40 mM ATP(ATP的Km),在室温下孵育60分钟。通过加入含有30μL SA-APC和4 nM PT-66抗体的30 mM EDTA停止反应,并在Perkin Elmer Envision上测量平板。使用四参数线性回归算法确定试验化合物的IC50值。 DiscoveRx Screening[1] 在KINOMEscan测定中,化合物在10μM下进行筛选,主要筛选结合相互作用的结果报告为“对照百分比”,其中较低的数字表示基质中的命中率较高。>35%的值被认为是“无命中”。Kd测定在DiscoveRx进行评估。 竞争性蛋白结合试验[1] 将含有10%胎牛血清(CCM)的人血浆和细胞培养基以2μM的终浓度掺入受试化合物。将加标血浆(1 mL)和CCM(1 mL)放入组装好的透析细胞的相对侧,用半透膜隔开。透析细胞在37°C水浴中缓慢旋转达到平衡所需的时间。测量透析后血浆和CCM质量,并用LC/MS/MS测定血浆和CCM中的试验化合物浓度。 代谢稳定性[1] 使用合并的肝微粒体组分(最终蛋白质浓度为0.5mg/mL)在3μM的最终试验化合物浓度下测定体外代谢稳定性。通过加入NADPH再生系统引发反应。在不同时间点将25μL的反应混合物等分试样转移到含有淬火溶液的板上。用LC/MS/MS测定反应混合物中的试验化合物浓度。使用充分搅拌的肝脏模型计算预测的清除率,不受蛋白质限制。 还使用氚化测试化合物在冷冻保存的肝细胞中测定了代谢稳定性。孵育混合物含有1×106个肝细胞/mL和1μM氚化试验化合物(2.5μCi)。在37°C的温度下,在95%空气/5%二氧化碳(v/v)的潮湿环境中轻轻摇晃进行孵化。在0、1、3和6小时后取出50μL的等分试样,并将其加入100μL的淬火溶液中。在与HPLC系统耦合的流动闪烁无线电探测器上分析样品。以无细胞对照样品作为参考,根据放射性检测器的峰面积对代谢物进行定量。通过测量所形成的放射性标记代谢物和试验化合物的总峰面积的ta百分比,来确定肝细胞中的代谢稳定性。 |
||
| 细胞实验 |
细胞交叉筛选活性测定[1]
将骨髓来源的小鼠肥大细胞(BMMC)、HUVEC或SK-N-MCs以(1-2)×106个细胞/孔的速度重新悬浮在Tyrode's缓冲液(BMMCs)或RPMI中,与化合物稀释液一起孵育1小时,然后用50 ng/mL SCF(BMMC)、50 ng/mL VEGF(HUVECs)或100 ng/mL GDNF(SK-N-MC)刺激。刺激3-15分钟后,将细胞在PBS中洗涤并重新悬浮在细胞裂解缓冲液中,通过SDS-PAGE解析蛋白质。对BMMC中的磷酸化cKit进行免疫检测评估,并归一化为总PLCγ2、HUVEC中的磷酸KDR和SK-N-MCs中的磷酸Ret。通过使用红外结合的二抗和Odyssey软件实现了检测。 Jak2活性测定[1] TF1细胞在1%胎牛血清(FBS)RPMI培养基中以1×106个细胞/mL的浓度饥饿过夜。将细胞重新悬浮在新鲜的无血清RPMI中,用复合稀释液孵育1小时,然后用5单位/mL促红细胞生成素刺激。将细胞溶解在50μL RIPA缓冲液中,使用MSD磷酸化Stat5定量板检测磷酸化Stat5。 MV-4-11增殖试验[1] 通过测量化合物对MV-4-11细胞增殖的抑制作用来确定对细胞Flt3活性的功能影响。在96孔平底组织培养板中,将总共104个细胞在含有10%FBS的RPMI培养基中稀释,并在37°C下与复合稀释液一起孵育72小时。将Alamar蓝(10%)加入细胞中,在37°C下再孵育12-18小时,通过570/600 nm的分光光度计读数确定相对细胞数量的抑制。 拉莫斯试验(pBLNK)[1] 在直立T175 Falcon TC烧瓶中,Ramos细胞在无血清RPMI中以2×106个细胞/mL的浓度饥饿1小时。将细胞离心(1100 rpm,5分钟),在37°C下,在3倍系列稀释的试验化合物或DMSO对照中以5×106个细胞/mL的密度孵育1小时。在37°C下用3μg/mL抗人IgM F(ab)2孵育5分钟,刺激细胞。将细胞制成颗粒,并在50μL细胞裂解缓冲液中裂解。使用涂有30ng/孔总BLNK捕获抗体的MSD高结合板检测磷酸BLNK 1小时。加入裂解物,用TBS-1%吐温-20洗涤细胞,并用抗磷-Blnk-Y96抗体检测。与对照孔相比,定量pBLNK的抑制作用。 人类B细胞增殖[1] 将分离的人B细胞在37°C水浴中解冻,并在含有10%FBS、100单位/mL青霉素-链霉素、0.01 M HEPES、2 mM GlutaMAX、5 mM丙酮酸钠和10 mMβ-巯基乙醇的RPMI 1640培养基中放置5小时,然后按照制造商的说明装入5μM CFSE。将圆底96孔板中的细胞(3×105个细胞/200μL/孔)与化合物在37°C培养箱中孵育1小时,然后用20μg/mL山羊F(ab′)2抗人IgM和20μg/mL小鼠抗CD40刺激,并在37°C培养箱中培养90小时。将细胞在PBS+4%FBS中漂洗一次,并在冰上用7AAD孵育30分钟。将细胞以300g的剂量制粒10分钟,冲洗两次,并在7AAD群体上通过流式细胞术进行分析,根据荧光素染色的减少来估计增殖情况。 免疫复合物刺激TNFα的产生[1] 将冷冻的人单核细胞在37°C水浴中快速解冻,并在RPMI 1640培养基中于37°C下静置3小时,该培养基补充了10%热灭活FBS、2 mM谷氨酸、1×丙酮酸钠、0.1 M HEPES、10 mMβ-巯基乙醇和100单位/mL青霉素-链霉素,然后以1×105个细胞/孔的速度在100μL完全RPMI的96孔板中铺板。将细胞与化合物一起孵育1小时,并在37°C下用4μL 40μg/mL的免疫复合物(300μL多克隆山羊F(ab′)2抗人Fc储备溶液+35μL纯化的人IgG+65μL培养基(最终质量比为3:1)在冰上孵育1小时时)刺激16小时。收集培养上清液并将其储存在-20°C下,直到使用单重Meso Scale TNFα试剂盒分析TNFα水平。 CD63全血检测[1] 新鲜的人全血在肝素钠真空采血管中采集。将2×系列稀释的2μL化合物样品加入96孔微量滴定板中的100μL全血中,在37°C下孵育1小时。在37°C下加入20μL抗人IL-3增强缓冲液B样品10分钟,然后在37°C下刺激山羊抗人IgE 20分钟。将反应物置于冰上以阻止脱颗粒,并用20μL的抗CD63-FITC/抗CD123-PE/抗HLA-DR PerCEP对细胞进行染色。用1.6 mL缓冲液G裂解红细胞10分钟并避光,在室温下以1300 rpm离心10分钟,收获细胞颗粒。用1.0 mL洗涤缓冲液A洗涤颗粒一次5分钟,然后重新离心。通过Canto FACS Calibur上的荧光激活细胞分选(FACS)分析来测量CD123+/HLA-细胞上的CD63表达,并使用CD63表达(%)与DMSO对照来确定全血中的EC50。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
The in vitro ADME and in vivo PK properties of Entospletinib (GS9973) are summarized in Table 5. Entospletinib (GS9973) is highly protein bound across species, with a human free fraction of 2.7%. Entospletinib (GS9973) was relatively stable in human liver microsomes (predicted Cl = 0.29 L/h/kg) but was less stable in preclinical species. In vivo, the clearance relative to hepatic blood flow was low in rat but was higher in dog, with dog having a relatively lower extent of protein binding. Entospletinib (GS9973) showed good bidirectional permeability across Caco-2 cell monolayers, indicating good absorption potential and low potential for efflux at concentrations likely to be achieved clinically. When dosed orally at 1 mg/kg in solution, Entospletinib (GS9973) showed moderate to high bioavailability in rat and dog. A comparison of the bioavailability and hepatic extraction in these species indicates that the extent of absorption from the GI tract is high (>75%). Since inhibition of metabolizing enzymes has the potential to cause clinically relevant drug–drug interactions, we evaluated the ability of Entospletinib (GS9973) to inhibit CYP1A2, 2C9, 2C19, 2D6, and 3A4. IC50 values were >10 μM in all cases. The solubility of crystalline Entospletinib (GS9973) in simulated intestinal fluid under both fed and fasted conditions was quite low (16 and 2 μM, respectively), most likely due to its high crystallinity and melting point (326 °C).[1]
|
||
| 参考文献 |
|
||
| 其他信息 |
Entospletinib has been used in trials studying the treatment of Oncology, Follicular Lymphoma, B-cell Malignancies, Mantle Cell Lymphoma, and Non-Hodgkin Lymphoma, among others.
Entospletinib is an orally available inhibitor of spleen tyrosine kinase (Syk), with potential antineoplastic activity. Upon oral administration of entospletinib, this agent may inhibit the activity of Syk, which inhibits B-cell receptor (BCR) signaling and leads to an inhibition of tumor cell activation, migration, adhesion and proliferation. Syk, a non-receptor cytoplasmic, BCR-associated tyrosine kinase, is expressed in hematopoietic tissues and is often overexpressed in hematopoeitic malignancies. Drug Indication Treatment of acute myeloid leukaemia. Spleen tyrosine kinase (Syk) is an attractive drug target in autoimmune, inflammatory, and oncology disease indications. The most advanced Syk inhibitor, R406, 1 (or its prodrug form fostamatinib, 2), has shown efficacy in multiple therapeutic indications, but its clinical progress has been hampered by dose-limiting adverse effects that have been attributed, at least in part, to the off-target activities of 1. It is expected that a more selective Syk inhibitor would provide a greater therapeutic window. Herein we report the discovery and optimization of a novel series of imidazo[1,2-a]pyrazine Syk inhibitors. This work culminated in the identification of GS-9973, 68, a highly selective and orally efficacious Syk inhibitor which is currently undergoing clinical evaluation for autoimmune and oncology indications.[1] Agents that target B-cell receptor (BCR) signaling in lymphoid malignancies including idelalisib (GS-1101) and fostamatinib which inhibit the delta isoform of PI3 kinase (PI3Kd) and spleen tyrosine kinase (Syk) respectively have shown significant clinical activity. By disrupting B-cell signaling pathways, idelalisib treatment has been associated with a dramatic lymph node response, but eradication of disease and relapse in high risk disease remain challenges. Targeting the BCR signaling pathway with simultaneous inhibition of PI3Kd and Syk has not yet been reported. We evaluated the pre-clinical activity of idelalisib combined with the novel and selective Syk inhibitor GS-9973 in primary peripheral blood and bone marrow Chronic Lymphocytic Leukemia (CLL) samples. Both PI3Kd and Syk inhibition reduced CLL survival and in combination induced synergistic growth inhibition and further disrupted chemokine signaling at nanomolar concentrations including in bone marrow derived and poor risk samples. Simultaneous targeting of these kinases may significantly increase clinical activity.[2] |
| 分子式 |
C23H21N7O
|
|---|---|
| 分子量 |
411.46
|
| 精确质量 |
411.18
|
| 元素分析 |
C, 67.14; H, 5.14; N, 23.83; O, 3.89
|
| CAS号 |
1229208-44-9
|
| 相关CAS号 |
1648797-46-9 (dimesylate);1229208-44-9;
|
| PubChem CID |
59473233
|
| 外观&性状 |
White to gray solid powder
|
| 密度 |
1.5±0.1 g/cm3
|
| 折射率 |
1.772
|
| LogP |
3.74
|
| tPSA |
86.6
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
595
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C, 67.14; H, 5.14; N, 23.83; O, 3.89
|
| InChi Key |
XSMSNFMDVXXHGJ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C23H21N7O/c1-2-17-14-25-28-20(17)13-16(1)21-15-30-8-7-24-23(30)22(27-21)26-18-3-5-19(6-4-18)29-9-11-31-12-10-29/h1-8,13-15H,9-12H2,(H,25,28)(H,26,27)
|
| 化学名 |
6-(1H-indazol-6-yl)-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine
|
| 别名 |
GS-9973; Entospletinib; GS9973; Entospletinib; 1229208-44-9; GS-9973; 6-(1H-Indazol-6-yl)-N-(4-morpholinophenyl)imidazo[1,2-a]pyrazin-8-amine; Entospletinib (GS-9973); Entospletinib [INN]; GS 9973;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.08 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 配方 2 中的溶解度: 4% DMSO+30% PEG 300+5% Tween 80+ddH2O:2.5mg/mL 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4304 mL | 12.1518 mL | 24.3037 mL | |
| 5 mM | 0.4861 mL | 2.4304 mL | 4.8607 mL | |
| 10 mM | 0.2430 mL | 1.2152 mL | 2.4304 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03010358 | Completed Has Results | Drug: Entospletinib Other: Laboratory Biomarker Analysis |
Anemia B-Cell Prolymphocytic Leukemia |
Alexey Danilov, MD | July 17, 2017 | Phase 1 Phase 2 |
| NCT02521376 | Completed Has Results | Drug: Entospletinib | Oncology | Gilead Sciences | November 16, 2015 | Phase 1 |
| NCT01796470 | Terminated Has Results | Drug: Entospletinib Drug: Idelalisib |
Chronic Lymphocytic Leukemia Mantle Cell Lymphoma |
Gilead Sciences | June 20, 2013 | Phase 2 |
| NCT05020665 | Terminated Has Results | Drug: Entospletinib Drug: Placebo |
Nucleophosmin 1-mutated Acute Myeloid Leukemia |
Kronos Bio | November 24, 2021 | Phase 3 |
Treatment of primary CLL cells (n =14) co-cultured with HS5 stromal cells with idelalisib (100 nM) or GS-9973 (100 nM), alone or in combination (100 nM each), results in decreased AKT phosphorylation.Oncotarget.2014 Feb 28;5(4):908-15. |
Disease status and biologic CLL disease characteristics.Oncotarget.2014 Feb 28;5(4):908-15. |
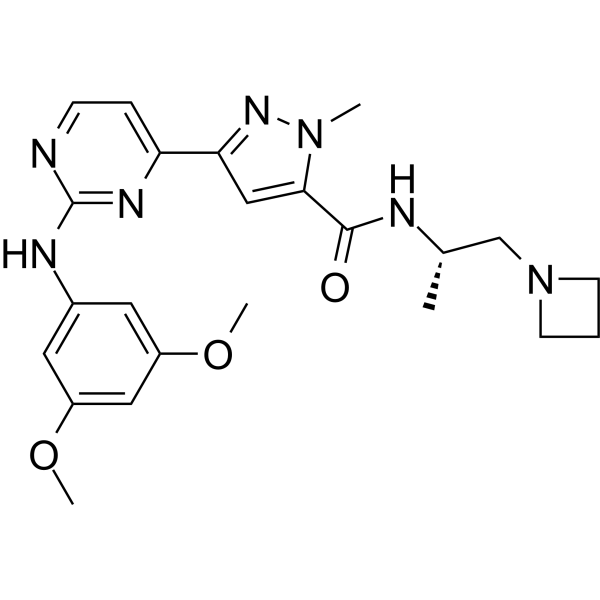 NMS-0963
NMS-0963
 Syk Inhibitor II hydrochloride
Syk Inhibitor II hydrochloride
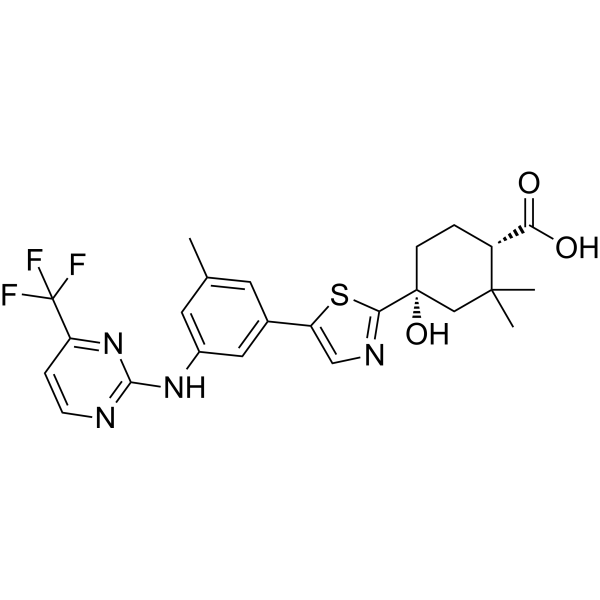 MK-8457
MK-8457
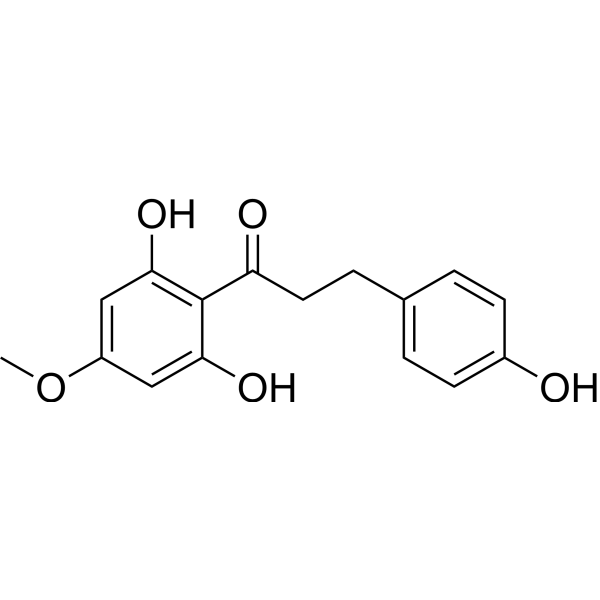 Asebogenin
Asebogenin
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
























 COA
COA
")
")
