| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Rac1 (IC50 = 1.1 μM)
|
|---|---|
| 体外研究 (In Vitro) |
本研究以已建立的Rac/Rac GEF抑制剂NSC23766的结构为基础,合成了一种新的Rac活性抑制剂EHop-016。在此,我们证明EHop-016抑制过表达Rac的MDA-MB-435转移癌细胞中的Rac活性,并表现出较高的内源性Rac活性。EHop-016对Rac的抑制IC50为1.1 μm,比NSC23766低约100倍。EHop-016对浓度≤5 μm的Rac1和Rac3具有特异性。在较高浓度下,EHop-016抑制相近的同源Cdc42。在显示高水平Rac GEF Vav2活性的MDA-MB-435细胞中,EHop-016抑制Vav2与无核苷酸的Rac1(G15A)的关联,G15A对活化的GEF具有高亲和力。EHop-016还抑制MDA-MB-231转移性乳腺癌细胞的Rac活性,并减少两种细胞系中Rac导向的板足形成。EHop-016降低Rac对PAK1 (p21活化激酶1)活性的下游作用和转移癌细胞的定向迁移。此外,在有效浓度(<5 μm)下,EHop-016不影响转化乳腺上皮细胞(MCF-10A)的活力,仅使MDA-MB-435细胞的活力降低20%。因此,EHop-016有望成为一种靶向治疗具有高Rac活性的转移性癌症的药物[1]。
体外活性:EHop-016 抑制 Rac 可通过补偿机制增加密切相关的 Rho GTPase RhoA 的活性。在 MDA-MB-435 细胞中,EHop-016 (2-5 μM) 抑制活性 Vav2 与 Rac1(G15A) 突变融合蛋白的结合,并降低 Rac 调节的细胞功能,包括板状伪足形成和细胞迁移。此外,EHop-016 还能抑制 MDA-MB-435 细胞的细胞活力,IC50 为 10 μM。 EHop-016 还抑制 KITD814V 诱导的 SM 和 AML 患者来源的细胞生长。 |
| 体内研究 (In Vivo) |
用 EHop-016 处理携带 KITD814V 的细胞可显着提高白血病小鼠的存活率。与上述体外研究结果一致,用EHop-016与NSC23766治疗kitd814v细胞显著提高白血病小鼠的存活率,脾脏大小和PB计数减少。[2]
|
| 酶活实验 |
Rac活性测定[1]
从MDA-MB-435和MDA-MB-231人转移癌细胞系(来自ATCC)的裂解物中测定Rac活性。将培养液(DMEM, 10% FBS, pH 7.5)中的癌细胞用载药(0.1% DMSO)或不同浓度的EHop-016 (0-10 μm)处理24小时。使用G-LISA Rac1激活检测试剂盒测定Rac1活性,方法如上所述。 为了生成每种抑制剂(ehopper -016或NSC23766)的IC50曲线,将三个独立重复实验的数据合并,并使用GraphPad Prism®的非线性回归函数拟合四参数剂量-反应曲线。 Rho GTPase活性测定[1] 用EHop-016处理MDA-MB-435和MDA-MB-231细胞裂解物24小时后,通过下拉法分析Rho、Rac和Cdc42活性。rhotekin的GST-Rho结合域用于分离活性gtp结合的Rho, GST-Cdc42和Rac相互结合(CRIB)域用于分离活性Rac- gtp或Cdc42- gtp,如上文所述。活性和总Rho gtpase用特异性抗体进行Western blotting鉴定。 Tiam-1 DH/PH结构域与Rac1(G15A)的相互作用[1] 将his标记的Tiam-1 DH-PH pET构建物转化到Rosetta DE3大肠杆菌细胞中,并使用His-Select镍亲和凝胶通过批量亲和层析纯化澄清的裂解物。Tiam-1用300 mm咪唑洗脱,用高效液相色谱柱Superdex 200分离。SDS-PAGE分析发现,在1.7 mg/ml时,Tiam-1的纯度大于95%。将GST-Rac1(G15A)谷胱甘肽琼脂糖或谷胱甘肽琼脂糖珠单独与不同浓度的EHop-016或NSC23766在裂解缓冲液(1% Igepal, 20 mm HEPES, 150 mm NaCl, 5 mm MgCl2, pH 7.5)中预孵育1小时。以2:1的Rac1(G15A)/Tiam-1浓度加入纯化的His-Tiam-1 DH/PH结构域,在4℃下孵育1h。拉下在1% Igepal缓冲液中洗涤三次,在HEPES缓冲液中洗涤一次,用抗his抗体进行Western印迹,可见His-Tiam-1 DH/PH结构域蛋白。 MDA-MB-435和MDA-MB-231人转移癌细胞系裂解物用于测定Rac活性。将在培养基(DMEM, 10% FBS, pH 7.5)中生长的癌细胞暴露于载体(0.1% DMSO)或不同浓度的EHop-016 (0-10 μM)中一整天。使用G-LISA Rac1激活测定试剂盒,测定Rac1活性。 |
| 细胞实验 |
荧光显微镜[1]
如前所述,将培养基中的MDA-MB-435或MDA-MB-231细胞用载药(0.1% DMSO)或EHop-016在2 μm和4 μm下处理24小时。将细胞固定,渗透,并用罗丹明phalloidin染色以显示F-actin。使用Spot数码相机在Olympus BX40荧光显微镜×600放大下获得荧光显微图。 细胞迁移试验[1] 如前所述,将静止的MDA-MB-435细胞用载体或不同浓度的EHop-016 (0-5 μm)处理24小时。将2 × 105个细胞置于Transwell室的上孔,下孔中含有10% FBS的培养基。在孵育4小时后,对每次处理后迁移到膜下的细胞数量进行量化。用碘化丙啶染色固定细胞,观察细胞核。对于每个处理(三个生物实验,每个实验两个技术重复),在Olympus CKX41倒置荧光显微镜×200放大下对20个显微镜视野中的细胞进行定量。 细胞活力测定[1] 如前所述,将MDA-MB-231、MDA-MB-435或MCF-10A乳腺上皮细胞(来自ATCC)在载具(0.1% DMSO)或不同浓度的EHop-016 (0-10 μm)中孵育24小时。根据制造商的说明,使用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四唑细胞存活和增殖试剂盒测量细胞活力。 将乳腺上皮细胞(MDA-MB-231、MDA-MB-435或MCF-10A)在0.1% DMSO或不同浓度的EHop-016 (0-10 μM)中培养24小时。按照制造商的说明,使用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四唑细胞存活和增殖试剂盒测定细胞活力。 |
| 动物实验 |
KITD814V-bearing mice.
2.5 μM KITD814V-bearing 32D cells with EHop-016 are administered by i.v. injection. Transplantation into C3H/HeJ mice was carried out by administering a single i.v. injection of 2 × 106 32D cells bearing WT KIT or KITD814V with or without RacN17 or PakK299R, or 1 × 106 KITD814V-bearing 32D cells cultured overnight with DMSO (vehicle), 25 μM NSC23766, or 2.5 μM EHop-016. Mice were harvested at the time of moribundity, and PB, femurs, spleen, lungs, and liver were collected for histopathological and flow cytometric analysis.[2] |
| 参考文献 | |
| 其他信息 |
The Rho GTPase Rac regulates actin cytoskeleton reorganization to form cell surface extensions (lamellipodia) required for cell migration/invasion during cancer metastasis. Rac hyperactivation and overexpression are associated with aggressive cancers; thus, interference of the interaction of Rac with its direct upstream activators, guanine nucleotide exchange factors (GEFs), is a viable strategy for inhibiting Rac activity. We synthesized EHop-016, a novel inhibitor of Rac activity, based on the structure of the established Rac/Rac GEF inhibitor NSC23766. Herein, we demonstrate that EHop-016 inhibits Rac activity in the MDA-MB-435 metastatic cancer cells that overexpress Rac and exhibits high endogenous Rac activity. The IC(50) of 1.1 μM for Rac inhibition by EHop-016 is ∼100-fold lower than for NSC23766. EHop-016 is specific for Rac1 and Rac3 at concentrations of ≤5 μM. At higher concentrations, EHop-016 inhibits the close homolog Cdc42. In MDA-MB-435 cells that demonstrate high active levels of the Rac GEF Vav2, EHop-016 inhibits the association of Vav2 with a nucleotide-free Rac1(G15A), which has a high affinity for activated GEFs. EHop-016 also inhibits the Rac activity of MDA-MB-231 metastatic breast cancer cells and reduces Rac-directed lamellipodia formation in both cell lines. EHop-016 decreases Rac downstream effects of PAK1 (p21-activated kinase 1) activity and directed migration of metastatic cancer cells. Moreover, at effective concentrations (<5 μM), EHop-016 does not affect the viability of transformed mammary epithelial cells (MCF-10A) and reduces viability of MDA-MB-435 cells by only 20%. Therefore, EHop-016 holds promise as a targeted therapeutic agent for the treatment of metastatic cancers with high Rac activity.[1]
An acquired somatic mutation at codon 816 in the KIT receptor tyrosine kinase is associated with poor prognosis in patients with systemic mastocytosis and acute myeloid leukemia (AML). Treatment of leukemic cells bearing this mutation with an allosteric inhibitor of p21-activated kinase (Pak) or its genetic inactivation results in growth repression due to enhanced apoptosis. Inhibition of the upstream effector Rac abrogates the oncogene-induced growth and activity of Pak. Although both Rac1 and Rac2 are constitutively activated via the guanine nucleotide exchange factor (GEF) Vav1, loss of Rac1 or Rac2 alone moderately corrected the growth of KIT-bearing leukemic cells, whereas the combined loss resulted in 75% growth repression. In vivo, the inhibition of Vav or Rac or Pak delayed the onset of myeloproliferative neoplasms (MPNs) and corrected the associated pathology in mice. To assess the role of Rac GEFs in oncogene-induced transformation, we used an inhibitor of Rac, EHop-016, which specifically targets Vav1 and found that EHop-016 was a potent inhibitor of human and murine leukemic cell growth. These studies identify Pak and Rac GTPases, including Vav1, as potential therapeutic targets in MPN and AML involving an oncogenic form of KIT.[2] |
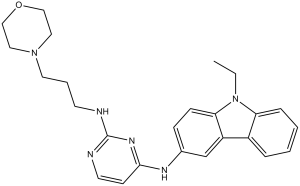
| 分子式 |
C25H30N6O
|
|
|---|---|---|
| 分子量 |
430.55
|
|
| 精确质量 |
430.248
|
|
| 元素分析 |
C, 69.74; H, 7.02; N, 19.52; O, 3.72
|
|
| CAS号 |
1380432-32-5
|
|
| 相关CAS号 |
|
|
| PubChem CID |
51031035
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| 密度 |
1.27
|
|
| LogP |
3.263
|
|
| tPSA |
73.7
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
8
|
|
| 重原子数目 |
32
|
|
| 分子复杂度/Complexity |
573
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O1C([H])([H])C([H])([H])N(C([H])([H])C1([H])[H])C([H])([H])C([H])([H])C([H])([H])N([H])C1=NC([H])=C([H])C(=N1)N([H])C1C([H])=C([H])C2=C(C=1[H])C1=C([H])C([H])=C([H])C([H])=C1N2C([H])([H])C([H])([H])[H]
|
|
| InChi Key |
AFTZZRFCMOAFCR-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C25H30N6O/c1-2-31-22-7-4-3-6-20(22)21-18-19(8-9-23(21)31)28-24-10-12-27-25(29-24)26-11-5-13-30-14-16-32-17-15-30/h3-4,6-10,12,18H,2,5,11,13-17H2,1H3,(H2,26,27,28,29)
|
|
| 化学名 |
4-N-(9-ethylcarbazol-3-yl)-2-N-(3-morpholin-4-ylpropyl)pyrimidine-2,4-diamine
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.81 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.81 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.81 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2% DMSO +30% PEG 300 +5% Tween 80 +ddH2O: 5mg/mL 配方 5 中的溶解度: 10 mg/mL (23.23 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3226 mL | 11.6131 mL | 23.2261 mL | |
| 5 mM | 0.4645 mL | 2.3226 mL | 4.6452 mL | |
| 10 mM | 0.2323 mL | 1.1613 mL | 2.3226 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 |
|---|
 |
 Synthesis and docking of EHop-016 into the putative GEF binding pocket of Rac1.J Biol Chem.2012 Apr 13;287(16):13228-38. |
 NSC 23766
NSC 23766
 K-Ras inhibitor 12
K-Ras inhibitor 12
 MLS-573151
MLS-573151
 EHT 1864 2HCl
EHT 1864 2HCl
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
