| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 10g |
|
||
| Other Sizes |
|

| 靶点 |
Tubulin; microtubule; microtubule depolymerization
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
多西他赛三水合物(RP-56976 三水合物)和葡磷酰胺 (GLU) 的单一和联合治疗均以剂量依赖性方式影响细胞活力。在 PC-3 和 LNCaP 细胞中,GLU 的 IC50 分别为 70±4 μM 和 86.8±8 μM。相反,在 PC-3 和 LNCaP 细胞中,单独使用多西他赛的 IC50 分别为 1.46±0.2 nM 和 3.08±0.4 nM。当GLU和多西紫杉醇共同治疗时,PC-3和LNCaP细胞中的IC50值分别降低至2.7±0.1 nM和0.75±0.3 nM [1]。 NCI-H460 针对西紫杉醇的半衰期在 72 小时后为 30 nM,在 24 小时后为 116 nM。根据 DTP 数据搜索的数据,在 NCI-60 细胞板上,多西紫杉醇的典型 IC50 为 14–34 nM [2]。
|
||
| 体内研究 (In Vivo) |
与 2-HALO 组相比,14 小时光照 (HALO) 组中多西他赛三水合物 (RP-56976 三水合物) 诱导的雌性小鼠肠细胞凋亡明显更高。接受多西他赛后,2-HALO组的Bax表达急剧增加,但14-HALO组则没有。相反,多西紫杉醇显着升高14-HALO组中裂解的Caspase-3的表达,但在2-HALO组中则没有。此外,多西紫杉醇显着降低了14-HALO组中生存素蛋白的表达,但在2-HALO组中没有显着降低。与用相同药物治疗的2-HALO组相比,用多西紫杉醇治疗的14-HALO组的生存素表达水平显着降低[3]。多西紫杉醇 (DOX) 以 7 mg/kg 的剂量静脉注射,而胡椒碱 (PIP) 以 3.5 mg/kg 的静脉推注剂量以及口服 35 mg/kg 和 3.5 mg/kg 的剂量给予。大鼠斯普拉格-戴利型。 Sprague-Dawley 大鼠同时口服 35 mg/kg PIP 和静脉推注 7 mg/kg 多西紫杉醇。当 PIP 和多西紫杉醇一起使用时,它们的体内暴露会协同增加 [4]。
|
||
| 酶活实验 |
体外微管蛋白聚合试验。[5]
如前所述制备微管蛋白。在含有1 mM GTP和1 mM 2-巯基乙醇的PEM缓冲液(pH 6.5、100 mM PIPES、2 mM EGTA和1 mM MgSO4)中,通过三个温度依赖性组装/拆卸循环分离猪脑微管蛋白。微管蛋白通过磷酸纤维素层析制备微管蛋白,并储存在-70°C下。在含有1mMGTP和5%甘油的PEM缓冲液(100 mM PIPES、1mMMgCl2和1mMEGTA)中,将微管蛋白与指定浓度的测试化合物(如多西他赛)混合。通过分光光度计在340nm处监测微管聚合。平台吸光度值用于计算[5]。
|
||
| 细胞实验 |
紫杉醇和多西他赛在非小细胞肺癌临床治疗中的广泛使用使得有必要找到生物标志物来识别可以从紫杉醇或多西他赛中受益的患者。在本研究中,将对紫杉醇和多西他赛具有不同敏感性的非小细胞肺癌细胞系NCI-H460应用于紫杉醇或多西他赛低剂量治疗不同时间点的DNA微阵列表达谱分析。并鉴定了调节药物反应的复杂信号通路,并对几种新的与敏感性相关的标记物进行了生物计算。反应基因的动态变化表明,紫杉醇的作用是急性的,但多西他赛的作用在NCI-H460细胞中至少持续48小时。对表达改变的基因进行功能注释表明,对这两种药物有反应的基因/途径存在显著差异。紫杉醇治疗诱导的基因表达变化主要富集在肌动蛋白细胞骨架(ACTC1、MYL2和MYH2)、酪氨酸蛋白激酶(ERRB4、KIT和TIE1)和局灶性粘附途径(MYL2、IGF1和FLT1)中,而对多西他赛反应的表达变化与细胞表面受体连接的信号转导(SHH、DRD5和ADM2)、细胞因子-细胞因子-受体相互作用(IL1A和IL6)和细胞周期调控(CCNB1、CCNE2和PCNA)高度相关。此外,我们还通过实时PCR证实了一些不同的表达模式。我们的研究将为紫杉醇和多西他赛选择疗法的临床应用提供潜在的生物标志物[2]。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption
The pharmacokinetic profile of docetaxel is consistent with a three-compartment model. The initial rapid decline represents the distribution to the peripheral compartments, and the late (terminal) phase is partly due to a relatively slow efflux of docetaxel from the peripheral compartment. The area under the curve (AUC) was dose proportional at doses between 70 mg/m2 and 115 mg/m2 with infusion times of 1 to 2 hours. In a group of patients with solid tumors given 100 mg/m2 of docetaxel intravenously, the Cmax and AUC were 2.41 μg/mL and 5.93 μg⋅h/mL, respectively. Route of Elimination Docetaxel was eliminated in urine and feces following oxidative metabolism of the tert-butyl ester group, but fecal excretion was the main elimination route. Within 7 days, urinary and fecal excretion accounted for approximately 6% and 75% of the administered radioactivity, respectively. In the first 48 hours, approximately 80% of the radioactivity recovered was excreted in feces. One major and three minor metabolites were excreted at this point, with less than 8% as the unchanged drug. Volume of Distribution Docetaxel has a steady-state volume of distribution of 113 L. Its pharmacokinetic profile is consistent with a three-compartment pharmacokinetic model. Clearance After the administration of 20–115 mg/m2 of intravenous docetaxel to cancer patients, the total body clearance was 21 L/h/m2. In patients aged 1 to 20 years with solid tumors that received 55 mg/m2 to 235 mg/m2 of docetaxel in a 1-hour intravenous infusion every 3 weeks, clearance was 17.3 L/h/m2. The initial rapid decline represents distribution to the peripheral compartments and the late (terminal) phase is due, in part, to a relatively slow efflux of docetaxel from the peripheral compartment. Mean steady state volume of distribution was 113 L. In vitro studies showed that docetaxel is about 94% protein bound, mainly to alpha1-acid glycoprotein, albumin, and lipoproteins. In three cancer patients, the in vitro binding to plasma proteins was found to be approximately 97%. Dexamethasone does not affect the protein binding of docetaxel. A study of (14)C-docetaxel was conducted in three cancer patients. Docetaxel was eliminated in both the urine and feces following oxidative metabolism of the tert-butyl ester group, but fecal excretion was the main elimination route. Within 7 days, urinary and fecal excretion accounted for approximately 6% and 75% of the administered radioactivity, respectively. About 80% of the radioactivity recovered in feces is excreted during the first 48 hours as 1 major and 3 minor metabolites with very small amounts (less than 8%) of unchanged drug. The pharmacokinetics of docetaxel have been evaluated in cancer patients after administration of 20 mg/m2 to 115 mg/sq m in phase 1 studies. The area under the curve (AUC) was dose proportional following doses of 70 mg/sq m to 115 mg/sq m with infusion times of 1 to 2 hours. Docetaxel's pharmacokinetic profile is consistent with a three-compartment pharmacokinetic model, with half-lives for the alpha, beta, and gamma phases of 4 min, 36 min, and 11.1 hr, respectively. Mean total body clearance was 21 L/hr/sq m. View More
Metabolism / Metabolites Biological Half-Life With plasma sampling up to 8 to 22 days after docetaxel infusion, the terminal elimination half-life was 116 hours. Doses between 70 and 115 mg/m2 with infusion times of 1 to 2 hours produce a triphasic elimination profile. The half-life of the alpha, beta, and gamma phases are 4 minutes, 36 minutes, and 11.1 hours, respectively. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation Most sources consider breastfeeding to be contraindicated during maternal antineoplastic drug therapy. No information is available on the clinical use of docetaxel during breastfeeding. It has been suggested that breastfeeding should be discontinued for 4 to 5 days after a dose, although the manufacturer recommends that breastfeeding be discontinued for 1 week after the last dose. Chemotherapy may adversely affect the normal microbiome and chemical makeup of breastmilk. Women who receive chemotherapy during pregnancy are more likely to have difficulty nursing their infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk A telephone follow-up study was conducted on 74 women who received cancer chemotherapy at one center during the second or third trimester of pregnancy to determine if they were successful at breastfeeding postpartum. Only 34% of the women were able to exclusively breastfeed their infants, and 66% of the women reported experiencing breastfeeding difficulties. This was in comparison to a 91% breastfeeding success rate in 22 other mothers diagnosed during pregnancy, but not treated with chemotherapy. Other statistically significant correlations included: (1) mothers with breastfeeding difficulties had an average of 5.5 cycles of chemotherapy compared with 3.8 cycles among mothers who had no difficulties; and (2) mothers with breastfeeding difficulties received their first cycle of chemotherapy on average 3.4 weeks earlier in pregnancy. Of the 9 women who received a taxane-containing regimen, 7 had breastfeeding difficulties. |
||
| 参考文献 |
|
||
| 其他信息 |
Docetaxel trihydrate is the trihydrate form of docetaxel. It is used for the treatment of breast, ovarian, and non-small cell lung cancer, and with prednisone or prednisolone in hormone-refractory metastatic prostate cancer. It has a role as an antineoplastic agent. It is a hydrate and a secondary alpha-hydroxy ketone. It contains a member of docetaxel anhydrous.
Docetaxel is a semi-synthetic, second-generation taxane derived from a compound found in the European yew tree, Taxus baccata. Docetaxel displays potent and broad antineoplastic properties; it binds to and stabilizes tubulin, thereby inhibiting microtubule disassembly which results in cell- cycle arrest at the G2/M phase and cell death. This agent also inhibits pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and displays immunomodulatory and pro-inflammatory properties by inducing various mediators of the inflammatory response. Docetaxel has been studied for use as a radiation-sensitizing agent. (NCI04) A semisynthetic analog of PACLITAXEL used in the treatment of locally advanced or metastatic BREAST NEOPLASMS and NON-SMALL CELL LUNG CANCER. See also: Docetaxel (annotation moved to). Drug Indication Breast cancer Taxespira in combination with doxorubicin and cyclophosphamide is indicated for the adjuvant treatment of patients with: operable node-positive breast cancer ; operable node-negative breast cancer . For patients with operable node-negative breast cancer , adjuvant treatment should be restricted to patients eligible to receive chemotherapy according to internationally established criteria for primary therapy of early breast cancer . Taxespira in combination with doxorubicin is indicated for the treatment of patients with locally advanced or metastatic breast cancer who have not previously received cytotoxic therapy for this condition. Taxespira monotherapy is indicated for the treatment of patients with locally advanced or metastatic breast cancer after failure of cytotoxic therapy. Previous chemotherapy should have included an anthracycline or an alkylating agent. Taxespira combination with trastuzumab is indicated for the treatment of patients with metastatic breast cancer whose tumours over express HER2 and who previously have not received chemotherapy for metastatic disease. Taxespira in combination with capecitabine is indicated for the treatment of patients with locally advanced or metastatic breast cancer after failure of cytotoxic chemotherapy. Previous therapy should have included an anthracycline. Non-small cell lung cancer Taxespira indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of prior chemotherapy. Taxespira in combination with cisplatin is indicated for the treatment of patients with unresectable, locally advanced or metastatic non-small cell lung cancer , in patients who have not previously received chemotherapy for this condition. Prostate cancer Taxespira in combination with prednisone or prednisolone is indicated for the treatment of patients with hormone refractory metastatic prostate cancer . Gastric adenocarcinoma Taxespira in combination with cisplatin and 5-fluorouracil is indicated for the treatment of patients with metastatic gastric adenocarcinoma, including adenocarcinoma of the gastroesophageal junction, who have not received prior chemotherapy for metastatic disease. Head and neck cancer Taxespira in combination with cisplatin and 5-fluorouracil is indicated for the induction treatment of patients with locally advanced squamous cell carcinoma of the head and neck. |
| 分子式 |
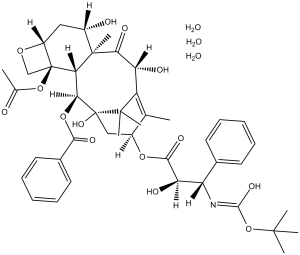
C43H53NO14.3H2O
|
|---|---|
| 分子量 |
861.93
|
| 精确质量 |
861.378
|
| 元素分析 |
C, 59.92; H, 6.90; N, 1.63; O, 31.55
|
| CAS号 |
148408-66-6
|
| 相关CAS号 |
Docetaxel;114977-28-5;Docetaxel-d5 trihydrate
|
| PubChem CID |
148123
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.37 g/cm3
|
| 沸点 |
1016.9ºC at 760 mmHg
|
| 熔点 |
186-192ºC
|
| 闪点 |
568.8ºC
|
| 蒸汽压 |
0mmHg at 25°C
|
| LogP |
3.457
|
| tPSA |
252.14
|
| 氢键供体(HBD)数目 |
8
|
| 氢键受体(HBA)数目 |
17
|
| 可旋转键数目(RBC) |
13
|
| 重原子数目 |
61
|
| 分子复杂度/Complexity |
1660
|
| 定义原子立体中心数目 |
11
|
| SMILES |
CC1=C2[C@H](C(=O)[C@@]3([C@H](C[C@@H]4[C@]([C@H]3[C@@H]([C@@](C2(C)C)(C[C@@H]1OC(=O)[C@@H]([C@H](C5=CC=CC=C5)NC(=O)OC(C)(C)C)O)O)OC(=O)C6=CC=CC=C6)(CO4)OC(=O)C)O)C)O.O.O.O
|
| InChi Key |
XCDIRYDKECHIPE-QHEQPUDQSA-N
|
| InChi Code |
InChI=1S/C43H53NO14.3H2O/c1-22-26(55-37(51)32(48)30(24-15-11-9-12-16-24)44-38(52)58-39(3,4)5)20-43(53)35(56-36(50)25-17-13-10-14-18-25)33-41(8,34(49)31(47)29(22)40(43,6)7)27(46)19-28-42(33,21-54-28)57-23(2)45;;;/h9-18,26-28,30-33,35,46-48,53H,19-21H2,1-8H3,(H,44,52);3*1H2/t26-,27-,28+,30-,31+,32+,33-,35-,41+,42-,43+;;;/m0.../s1
|
| 化学名 |
[(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4-acetyloxy-1,9,12-trihydroxy-15-[(2R,3S)-2-hydroxy-3-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]oxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate;trihydrate
|
| 别名 |
RP56976 (NSC 628503) Trihydrate; RP56976; NSC 628503; RP-56976; NSC628503; RP 56976; Docetaxel hydrate; XRP6976; Docetaxel (Trihydrate); Docetaxel (as trihydrate); RP 56976; CHEBI:59809; NSC-628503; Docetaxel trihydrate, Trade name: Taxotere.
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (2.90 mM) (饱和度未知) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL 澄清 EtOH 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (2.90 mM) (饱和度未知) in 10% EtOH + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清乙醇储备液加入 900 μL 20% SBE-β-CD 生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (2.90 mM) (饱和度未知) in 10% EtOH + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.08 mg/mL (2.41 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清的DMSO储备液加入400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 5 中的溶解度: ≥ 2.08 mg/mL (2.41 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100μL 20.8mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 6 中的溶解度: ≥ 2.08 mg/mL (2.41 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.1602 mL | 5.8009 mL | 11.6019 mL | |
| 5 mM | 0.2320 mL | 1.1602 mL | 2.3204 mL | |
| 10 mM | 0.1160 mL | 0.5801 mL | 1.1602 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA






