
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| Other Sizes |
|

| 靶点 |
Glucocorticoid receptor
|
|---|---|
| 体外研究 (In Vitro) |
地塞米松,也称为十六醇,作为多种转录因子的调节剂,包括核因子-AT、核因子-kB 和激活蛋白-1,进而激活和抑制与炎症反应相关的重要基因 [1] 。地塞米松的 EC50 为 2.2 nM,可有效阻止 A549 细胞释放粒细胞巨噬细胞集落刺激因子 (GM-CSF)。当剂量大于抑制 GM-CSF 产生的剂量时,地塞米松 (EC50=36 nM) 显示与糖皮质激素受体 (GR) DNA 结合相关,发生率高 10-100 倍。它还促进β2受体转录。 GM-CSF 释放的抑制与地塞米松对 3×κB(NF-κB、IκBα 和 I-κBβ)的抑制有关 (IC50=0.5 nM) [2]。
|
| 体内研究 (In Vivo) |
地塞米松可用于创建动物神经损伤、肌肉萎缩和线粒体疾病模型。先前已有文献报道,用 2 × 5 mg/kg 剂量的地塞米松 (Hexadecadrol) 治疗可有效抑制脂多糖 (LPS) 诱导的炎症。在我们的实验系统中,与暴露于脂多糖(LPS)并单独注射载体(盐水)的小鼠相比,单剂量地塞米松 10 mg/kg(腹腔注射)治疗显着减少了粒细胞的募集和氧自由基的自发产生。在吸入 LPS 前一小时和吸入后一小时时,效果具有统计学意义。通过施用水雾剂,BALF 中的粒细胞数量降低至与健康动物相似的水平 [3]。地塞米松治疗组的老鼠比对照组的老鼠吃得更少,体重也更轻。尽管吃同样量的食物,接受治疗的老鼠的体重却比成对喂养的老鼠轻。注射地塞米松五天后,肝脏质量(+42%)和肝脏与体重之比(+65%)显着增加。治疗五天后,腓肠肌湿重下降了20%,但与体重的关系(g/100 g体重)没有变化,表明体重减轻和肌肉减重是同步的[4] 。
|
| 酶活实验 |
1.糖皮质激素在控制哮喘和类风湿性关节炎等慢性炎症性疾病方面非常有效,但其抗炎作用的确切分子机制尚不清楚。它们通过与细胞质受体(GR)结合来激活或抑制基因表达。这可能是通过GR与DNA的直接结合(反式激活)或通过抑制AP-1和NF-kappaB等转录因子的活性(反式抑制)发生的。2.局部活性类固醇丙酸氟替卡松(EC50=1.8 x 10(-11)M)和布地奈德(EC50=5.0 x 10(-11)M)在抑制A549细胞释放GM-CSF方面比替普瑞定(EC50=8.3 x 10(-10)M)、布替西科特(EC50=3.7 x 10(-8)M)以及地塞米松(EC50=2.2 x 10(-9)M)更有效。抗糖皮质激素RU486还抑制了这些细胞中GM-CSF的释放(IC50=1.8 x 10(-10)M)。3.发现丙酸氟替卡松(EC50=9.8 x 10(-10)M)、布地奈德(EC50=1.1 x 10(-9)M)和地塞米松(EC50=3.6 x 10(-8)M)诱导β2受体转录的浓度依赖性能力与GR DNA结合有关,其浓度比抑制GM-CSF释放高10-100倍。24小时后,未观察到NF-kappaB、IkappaBalpha或I-kappaBbeta内源性抑制剂的诱导,糖皮质激素未改变IL-1β降解并随后诱导IkappaB的能力。4.丙酸氟替卡松(IC50=0.5 x 10(-11)M)、布地奈德(IC50=2.7 x 10(-11M))、地塞米松(IC50=0.5 x 10(-9)M)和RU486(IC50=2.7 x 10-11)抑制3 xκB的能力与抑制GM-CSF释放有关。5.这些数据表明,一系列糖皮质激素的抗炎特性与其反式加压而非反式激活基因的能力有关[2]。
|
| 细胞实验 |
糖皮质激素是一种广泛应用于临床实践的抗炎药。越来越多的证据表明外泌体是炎症的重要介质,但糖皮质激素是否调节外泌体的分泌和功能尚不清楚。在本研究中,我们观察到经地塞米松处理后,脂多糖(LPS)诱导的RAW264.7巨噬细胞的外泌体分泌减少。重要的是,从LPS诱导的RAW264.7巨噬细胞中分离的外泌体增加了RAW264.6细胞中TNF-α和IL-6的产生。然而,在用从地塞米松处理的细胞中分离的外泌体治疗后,这种增加不太明显。此外,地塞米松降低了LPS诱导的RAW264.7巨噬细胞外泌体中促炎微小RNA-155的表达。我们假设外泌体是糖皮质激素在LPS诱导的巨噬细胞炎症反应中抗炎作用的新靶点。这些发现将有利于抗炎治疗新方法的开发[7]。
|
| 动物实验 |
Synthetic glucocorticoids are pharmaceutical compounds prescribed in human and veterinary medicine as anti-inflammatory agents and have the potential to contaminate natural watersheds via inputs from wastewater treatment facilities and confined animal-feeding operations. Despite this, few studies have examined the effects of this class of chemicals on aquatic vertebrates. To generate data to assess potential risk to the aquatic environment, we used fathead minnow 21-d reproduction and 29-d embryo-larvae assays to determine reproductive toxicity and early-life-stage effects of Dexamethasone. Exposure to 500 µg Dexamethasone/L in the 21-d test caused reductions in fathead minnow fecundity and female plasma estradiol concentrations and increased the occurrence of abnormally hatched fry. Female fish exposed to 500 µg dexamethasone/L also displayed a significant increase in plasma vitellogenin protein levels, possibly because of decreased spawning. A decrease in vitellogenin messenger ribonucleic acid (mRNA) expression in liver tissue from females exposed to the high dexamethasone concentration lends support to this hypothesis. Histological results indicate that a 29-d embryo-larval exposure to 500 µg dexamethasone/L caused a significant increase in deformed gill opercula. Fry exposed to 500 µg dexamethasone/L for 29 d also exhibited a significant reduction in weight and length compared with control fry. Taken together, these results indicate that nonlethal concentrations of a model glucocorticoid receptor agonist can impair fish reproduction, growth, and development.[1]
For vaccination, RNA was formulated with liposomes consisting of DOTMA and DOPE at a charge ratio (+):(-) of 1.3:2 yielding negatively charged RNA-Lipoplexes (RNA-LPX) as described previously.23 RNA-LPX comprising 20 µg gp70 RNA, 15 µg Reps1 and Adpgk RNA each or 30 µg eGFP RNA was injected i.v. in C57BL/6 or BALB/c mice as described in Figures 1 and 6(a). If not otherwise stated, Dexamethasone was injected i.p. at a dose of 4 mg/kg in 200 µL PBS. For lung metastasis experiments, 5 × 105 CT26 cells were injected i.v. in 100 µL PBS and treatment with 40 µg gp70 RNA-LPX and Dexa was performed as depicted in Figure 5(a). CT26 lung tumor burden was quantified after tracheal ink (1:10 diluted in PBS) injection and fixation with Fekete’s solution (5 ml 70% ethanol, 0.5 ml formalin, and 0.25 ml glacial acetic acid). A total of 1 × 105 MC38 tumor cells was implanted subcutaneously in the right hind flank in 100 µL of HBSS + matrigel. MC38 tumor-bearing mice were vaccinated with 50 µg RNA-LPX and treated with 4 mg/kg Dexamethasone as depicted in Figure 5(e), 5 mg/kg Methylprednisolone (Pfizer) or with 0.25 mg/kg Dexamethasone 5 minutes post-vaccination. Tumors were monitored at least twice per week and mice were euthanized if tumors became ulcerated or exceeded the acceptable size limit of 2000 mm3 according to IACUC.[8] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Absorption via the intramuscular route is slower than via the intravenous route. A 3mg intramuscular dose reaches a Cmax of 34.6±6.0ng/mL with a Tmax of 2.0±1.2h and an AUC of 113±38ng\*h/mL. A 1.5mg oral dose reaches a Cmax of 13.9±6.8ng/mL with a Tmax of 2.0±0.5h and an AUC of 331±50ng\*h/mL. Oral dexamethasone is approximately 70-78% bioavailable in healthy subjects. Corticosteroids are generally eliminated predominantly in the urine. However, dexamethasone is <10% elminated in urine. A 1.5mg oral dose of dexamethasone has a volume of distribution of 51.0L, while a 3mg intramuscular dose has a volume of distribution of 96.0L. A 20mg oral tablet has a clearance of 15.7L/h. A 1.5mg oral dose of dexamethasone has a clearance of 15.6±4.9L/h while a 3.0mg intramuscular dose has a clearance of 9.9±1.4L/h. Absorbed into aqueous humor, cornea, iris, choroid ciliary body, and retina. Systemic absorption occurs, but may be significant only at higher dosages or in extended pediatric therapy. /Corticosteroids (Ophthalmic)/ Dogs (mixed-breed) were administered dexamethasone alcohol or dexamethasone 21-isonicotinate as a solution iv or im (1 mg/kg bw), or dexamethasone 21-isonicotinate as a suspension im (0.1 or 1 mg/kg bw). Plasma concentrations were determined with HPLC up to 120 hours after treatment. The elimination half-life after iv administration was 120-140 minutes for both formulations. Following im administration, absorption was rapid with peak plasma concentrations at 30-40 minutes for both solutions. Bioavailability after im administration was 100% for dexamethasone alcohol but 40% for dexamethasone 21-isonicotinate. After im administration of dexamethasone 21-isonicotinate as a suspension, dexamethasone was not detected in plasma, suggesting a long absorption phase Crl:SD(CD)BR rats were administered a single im dose of 9 ug, (1,2,4-3H)-dexamethasone/kg bw. Radioactivity was measured up to 96 hours after administration in plasma (pre- and post-freeze dried), urine, feces and expired air. Tritium exchange was measured in stored urine. Highest plasma levels were observed 6 hours after dosing (3.7 ug equivalents/g), declining rapidly thereafter to 0.15 ug equivalents/g. Within 24 hours 41% of the radioactivity was excreted in the urine. After 96 hours a mean of 44% of the radio-activity was excreted. Tritium exchange was observed both in plasma and urine. Following freeze-drying, the mean loss of radioactivity 96 hours after dosing was 87% and 37% in plasma and urine, respectively Male Wistar albino rats were administered 0.23 umol (1,2-3H) dexamethasone/kg bw, ip. Urine and feces were collected up to 4 days after treatment. Within 96 hours 74% of the dose was excreted, 30% in the urine and 44% in the feces For more Absorption, Distribution and Excretion (Complete) data for DEXAMETHASONE (11 total), please visit the HSDB record page. Metabolism / Metabolites Dexamethasone is 6-hydroxylated by CYP3A4 to 6α- and 6β-hydroxydexamethasone. Dexamethasone is reversibly metabolized to 11-dehydrodexamethasone by corticosteroid 11-beta-dehydrogenase isozyme 2 and can also be converted back to dexamethasone by Corticosteroid 11-beta-dehydrogenase isozyme 1. Male Wistar albino rats were administered (3)H-dexamethasone orally at a dose of 1.14 nmol/kg bw. Thirty-one percent of the administered radioactivity was excreted in the urine within 4 days (most of it within the first 24 hours) as unconjugated metabolites. Unchanged dexamethasone accounted for 14%, 6-hydroxydexamethasone for 7.4%, and 20-dihydrodexamethasone for 1.1% of the urine radioactivity. In the urine of rats administered 0.23 umol/kg bw (1,2-3H)- dexamethasone ip, 10% of the administered radioactivity was associated with one polar metabolite of dexamethasone, likely to be 6-hydroxy-dexamethasone No parent compound could be detected in urine of patients after oral administration of a small dose of dexamethasone (<4 mg/day) for a few weeks. However, 60% was recovered as 6-beta-hydroxy-dexamethasone and 5-10% as 6-beta-hydroxy-20-dihydrodexamethasone. After the administration of about 15 mg dexamethasone/day metabolism occurred by an additional route involving epoxidation and subsequent hydrolysis, resulting in glycol formation in ring A Dexamethasone has known human metabolites that include 6-beta-OH DEXAMETHASONE and 6-alpha-OH DEXAMETHASONE. Hepatic. Biological Half-Life The mean terminal half life of a 20mg oral tablet is 4 hours. A 1.5mg oral dose of dexamethasone has a half life of 6.6±4.3h, while a 3mg intramuscular dose has a half life of 4.2±1.2h. 190 minutes (plasma) /Dexamethasone sodium phosphate/ Dogs (mixed-breed) were administered dexamethasone alcohol or dexamethasone 21-isonicotinate as a solution iv or im (1 mg/kg bw), or dexamethasone 21-isonicotinate as a suspension im (0.1 or 1 mg/kg bw). Plasma concentrations were determined with HPLC up to 120 hours after treatment. The elimination half-life after iv administration was 120-140 minutes for both formulations. |
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation Topical dexamethasone has not been studied during breastfeeding. Since only extensive application of the most potent corticosteroids cause systemic effects in the mother, it is unlikely that short-term application of topical corticosteroids would pose a risk to the breastfed infant by passage into breastmilk. However, it would be prudent to use the least potent drug on the smallest area of skin possible. It is particularly important to ensure that the infant's skin does not come into direct contact with the areas of skin that have been treated. Current guidelines allow topical corticosteroids to be applied to the nipples just after nursing for eczema, with the nipples cleaned gently before nursing. Only water-miscible cream or gel products should be applied to the breast because ointments may expose the infant to high levels of mineral paraffins via licking. Because absorption from the eye is limited, ophthalmic dexamethasone, including ocular inserts, would not be expected to cause any adverse effects in breastfed infants. To substantially diminish the amount of drug that reaches the breastmilk after using eye drops, place pressure over the tear duct by the corner of the eye for 1 minute or more, then remove the excess solution with an absorbent tissue. ◉ Effects in Breastfed Infants Topical application of a corticosteroid with relatively high mineralocorticoid activity (isofluprednone acetate) to the mother's nipples resulted in prolonged QT interval, cushingoid appearance, severe hypertension, decreased growth and electrolyte abnormalities in her 2-month-old breastfed infant. The mother had used the cream since birth for painful nipples. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. ◉ Summary of Use during Lactation Because little information is available on the use of systemic dexamethasone during breastfeeding, an alternate corticosteroid may be preferred, especially while nursing a newborn or preterm infant. Local injections, such as for tendinitis, would not be expected to cause any adverse effects in breastfed infants. Medium to large doses of corticosteroids, including dexamethasone, given systemically or injected into joints or the breast have been reported to cause temporary reduction of lactation. See also Dexamethasone, Topical. ◉ Effects in Breastfed Infants None reported with any corticosteroid. ◉ Effects on Lactation and Breastmilk Dexamethasone can cause a decrease in basal serum prolactin and thyrotropin-releasing hormone stimulated serum prolactin increase in nonnursing women. Medium to large doses of corticosteroids given systemically or injected into joints or the breast have been reported to cause temporary reduction of lactation. A study of 46 women who delivered an infant before 34 weeks of gestation found that a course of another corticosteroid (betamethasone, 2 intramuscular injections of 11.4 mg of betamethasone 24 hours apart) given between 3 and 9 days before delivery resulted in delayed lactogenesis II and lower average milk volumes during the 10 days after delivery. Milk volume was not affected if the infant was delivered less than 3 days or more than 10 days after the mother received the corticosteroid. An equivalent dosage regimen of dexamethasone might have the same effect. A study of 87 pregnant women found that betamethasone given as above during pregnancy caused a premature stimulation of lactose secretion during pregnancy. Although the increase was statistically significant, the clinical importance appears to be minimal. An equivalent dosage regimen of dexamethasone might have the same effect. A woman with postpartum depression who was breastfeeding her 8-week-old infant was treated with endovascular embolization for a spinal-dural arteriovenous fistula. Following the procedure, she was treated with intravenous dexamethasone 4 mg every 8 hours for 5 days, followed by oral dexamethasone 12 mg daily in a tapering regimen. After stopping breastfeeding for 3 days after the procedure, she noted a decreased milk supply on restarting breastfeeding, and a complete cessation of milk production 11 days after the procedure. Several measures to increase milk including domperidone supply failed. Breastmilk production resumed 36 hours after discontinuing dexamethasone and reached normal production after 8 days. At hospital discharge, she was exclusively nursing her infant. Protein Binding Dexamethasone is approximately 77% protein bound in plasma. The majority of protein binding is with serum albumin. Dexamethasone does not significantly bind to corticosteroid binding protein. |
| 参考文献 |
[1]. LaLone CA, et al. Effects of a glucocorticoid receptor agonist, Dexamethasone, on fathead minnow reproduction, growth, and development. Environ Toxicol Chem. 2012 Mar;31(3):611-22.
[2]. Adcock IM, et al. Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: preferential targetting of NF-kappaB and lack of I-kappaB involvement. Br J Pharmacol. 1999 Jun;127(4):1003-11 [3]. Rocksén D, et al. Differential anti-inflammatory and anti-oxidative effects of Dexamethasone and N-acetylcysteine in endotoxin-induced lung inflammation. Clin Exp Immunol. 2000 Nov;122(2):249-56 [4]. Roussel D, et al. Dexamethasone treatment specifically increases the basal proton conductance of rat liver mitochondria. FEBS Lett. 2003 Apr 24;541(1-3):75-9. [5]. Ballabh P, et al. Neutrophil and monocyte adhesion molecules in bronchopulmonary dysplasia, and effects of corticosteroids. Arch Dis Child Fetal Neonatal Ed. 2004 Jan;89(1):F76-83. [6]. Heidi Ledford. et al. Coronavirus Breakthrough: Dexamethasone Is First Drug Shown to Save Lives. Nature. 2020 Jun 16. [7]. Yun Chen, et al. Glucocorticoids inhibit production of exosomes containing inflammatory microRNA-155 in lipopolysaccharide-induced macrophage inflammatory responses. Int J Clin Exp Pathol 2018;11(7):3391-3397. [8]. Dexamethasone premedication suppresses vaccine-induced immune responses against cancer. Oncoimmunology. 2020; 9(1): 1758004. |
| 其他信息 |
Dexamethazone is an odorless white to off-white crystalline powder with a slightly bitter taste. (NTP, 1992)
Dexamethasone is a fluorinated steroid that is 9-fluoropregna-1,4-diene substituted by hydroxy groups at positions 11, 17 and 21, a methyl group at position 16 and oxo groups at positions 3 and 20. It is a synthetic member of the class of glucocorticoids. It has a role as an adrenergic agent, an antiemetic, an antineoplastic agent, an environmental contaminant, a xenobiotic, an immunosuppressive agent and an anti-inflammatory drug. It is a fluorinated steroid, a 3-oxo-Delta(1),Delta(4)-steroid, a glucocorticoid, a 20-oxo steroid, an 11beta-hydroxy steroid, a 17alpha-hydroxy steroid and a 21-hydroxy steroid. It derives from a hydride of a pregnane. Dexamethasone, or MK-125, is a corticosteroid fluorinated at position 9 used to treat endocrine, rheumatic, collagen, dermatologic, allergic, ophthalmic, gastrointestinal, respiratory, hematologic, neoplastic, edematous, and other conditions. Developed in 1957, it is structurally similar to other corticosteroids like [hydrocortisone] and [prednisolone]. Dexamethasone was granted FDA approval on 30 October 1958. In a press release for the Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial on 16 June 2020, dexamethasone was recommended for use in COVID-19 patients with severe respiratory symptoms. Dexamethasone reduced deaths by approximately one third in patients requiring ventilation and by one fifth in those requiring oxygen. Dexamethasone is a Corticosteroid. The mechanism of action of dexamethasone is as a Corticosteroid Hormone Receptor Agonist. Dexamethasone has been reported in Aspergillus ochraceopetaliformis, Penicillium chrysogenum, and other organisms with data available. Dexamethasone is a synthetic adrenal corticosteroid with potent anti-inflammatory properties. In addition to binding to specific nuclear steroid receptors, dexamethasone also interferes with NF-kB activation and apoptotic pathways. This agent lacks the salt-retaining properties of other related adrenal hormones. (NCI04) An anti-inflammatory 9-fluoro-glucocorticoid. [PubChem] An anti-inflammatory 9-fluoro-glucocorticoid. See also: Dexamethasone Sodium Phosphate (narrower); Dexamethasone Isonicotinate (is active moiety of); Dexamethasone Dipropionate (is active moiety of) ... View More ... Drug Indication Dexamethasone and [ciprofloxacin] otic suspension is indicated for bacterial infections with inflammation in acute otitis media and acute otitis externa. Intramuscular and intravenous injections are indicated for a number of endocrine, rheumatic, collagen, dermatologic, allergic, ophthalmic, gastrointestinal, respiratory, hematologic, neoplastic, edematous, and other conditions. Oral tablets are indicated for the treatment of multiple myeloma. An intravitreal implant is indicated for some forms of macular edema and non-infectious posterior uveitis affecting the posterior of the eye. Various ophthalmic formulations are indicated for inflammatory conditions of the eye. FDA Label Ozurdex is indicated for the treatment of adult patients with macular oedema following either branch retinal-vein occlusion (BRVO) or central retinal-vein occlusion (CRVO). Ozurdex is indicated for the treatment of adult patients with inflammation of the posterior segment of the eye presenting as noninfectious uveitis. Ozurdex is indicated for the treatment of adult patients with visual impairment due to diabetic macular oedema (DME) who are pseudophakic or who are considered insufficiently responsive to, or unsuitable for non-corticosteroid therapy. Treatment of multiple myeloma. Treatment of postoperative pain and inflammation associated with ophthalmic surgery Treatment of diabetic macular oedema Chronic non-infectious intermediate or posterior uveitis Other retinal vascular occlusion Mechanism of Action The short term effects of corticosteroids are decreased vasodilation and permeability of capillaries, as well as decreased leukocyte migration to sites of inflammation. Corticosteroids binding to the glucocorticoid receptor mediates changes in gene expression that lead to multiple downstream effects over hours to days. Glucocorticoids inhibit neutrophil apoptosis and demargination; they inhibit phospholipase A2, which decreases the formation of arachidonic acid derivatives; they inhibit NF-Kappa B and other inflammatory transcription factors; they promote anti-inflammatory genes like interleukin-10. Lower doses of corticosteroids provide an anti-inflammatory effect, while higher doses are immunosuppressive. High doses of glucocorticoids for an extended period bind to the mineralocorticoid receptor, raising sodium levels and decreasing potassium levels. Corticosteroids diffuse across cell membranes and complex with specific cytoplasmic receptors. These complexes then enter the cell nucleus, bind to DNA, and stimulate transcription of mRNA and subsequent protein synthesis of enzymes ultimately responsible for anti-inflammatory effects of topical application of corticosteroids to the eye. In high concentrations which may be achieved after topical application, corticosteroids may exert direct membrane effects. Corticosteroids decrease cellular and fibrinous exudation and tissue infiltration, inhibit fibroblastic and collagen-forming activity, retard epithelial regeneration, diminish postinflammatory neovascularization and reduce toward normal levels the excessive permeability of inflamed capillaries. /Corticosteroids (Otic)/ Glucocorticoids are capable of suppressing the inflammatory process through numerous pathways. They interact with specific intracellular receptor proteins in target tissues to alter the expression of corticosteroid-responsive genes. Glucocorticoid-specific receptors in the cell cytoplasm bind with steroid ligands to form hormone-receptor complexes that eventually translocate to the cell nucleus. There these complexes bind to specific DNA sequences and alter their expression. The complexes may induce the transcription of mRNA leading to synthesis of new proteins. Such proteins include lipocortin, a protein known to inhibit PLA2a and thereby block the synthesis of prostaglandins, leukotrienes, and PAF. Glucocorticoids also inhibit the production of other mediators including AA metabolites such as COX, cytokines, the interleukins, adhesion molecules, and enzymes such as collagenase. /Glucocorticoids/ Corticosteroids diffuse across cell membranes and complex with specific cytoplasmic receptors. These complexes then enter the cell nucleus, bind to DNA (chromatin), and stimulate transcription of messenger RNA (mRNA) and subsequent protein synthesis of various inhibitory enzymes responsible for the anti-inflammatory effects of topical corticosteroids. These anti-inflammatory effects include inhibition of early processes such as edema, fibrin deposition, capillary dilatation, movement of phagocttes into the area, and phagocytic activities. Later processes, such as capillary production, collagen deposition, and keloid formation also are inhibited by corticosteroids. The overall actions of topical corticosteroids are catabolic. /Corticosteroids (topical)/ |
| 分子式 |
C22H29FO5
|
|
|---|---|---|
| 分子量 |
392.46
|
|
| 精确质量 |
392.199
|
|
| 元素分析 |
C, 67.33; H, 7.45; F, 4.84; O, 20.38
|
|
| CAS号 |
50-02-2
|
|
| 相关CAS号 |
3936-02-5 (metasulfobenzoate sodium);3800-84-8 (sodium succinate);1177-87-3 (acetate);150587-07-8 (beloxil); 132245-57-9 (cipecilate); 2265-64-7 (isonicotinate); 14899-36-6 (palmitate); 312-93-6 (phosphate); 2392-39-4 (sodium phosphate);
|
|
| PubChem CID |
5743
|
|
| 外观&性状 |
Crystals from ether
WHITE TO PRACTICALLY WHITE CRYSTALLINE POWDER |
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
568.2±50.0 °C at 760 mmHg
|
|
| 熔点 |
255-264ºC
|
|
| 闪点 |
297.5±30.1 °C
|
|
| 蒸汽压 |
0.0±3.5 mmHg at 25°C
|
|
| 折射率 |
1.592
|
|
| LogP |
1.87
|
|
| tPSA |
94.83
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
28
|
|
| 分子复杂度/Complexity |
805
|
|
| 定义原子立体中心数目 |
8
|
|
| SMILES |
F[C@@]12[C@]3(C=CC(C=C3CC[C@@]1([H])[C@]1([H])C[C@@H](C)[C@](O)(C(=O)CO)[C@]1(C[C@@H]2O)C)=O)C
|
|
| InChi Key |
UREBDLICKHMUKA-CXSFZGCWSA-N
|
|
| InChi Code |
InChI=1S/C22H29FO5/c1-12-8-16-15-5-4-13-9-14(25)6-7-19(13,2)21(15,23)17(26)10-20(16,3)22(12,28)18(27)11-24/h6-7,9,12,15-17,24,26,28H,4-5,8,10-11H2,1-3H3/t12-,15+,16+,17+,19+,20+,21+,22+/m1/s1
|
|
| 化学名 |
(8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-3-one
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.37 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
*20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.30 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (5.30 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.08 mg/mL (5.30 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 配方 5 中的溶解度: 30% PEG400+0.5% Tween80+5% Propylene glycol : 30mg/kg 配方 6 中的溶解度: 18.18 mg/mL (46.32 mM) in 0.5% CMC-Na/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5480 mL | 12.7402 mL | 25.4803 mL | |
| 5 mM | 0.5096 mL | 2.5480 mL | 5.0961 mL | |
| 10 mM | 0.2548 mL | 1.2740 mL | 2.5480 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
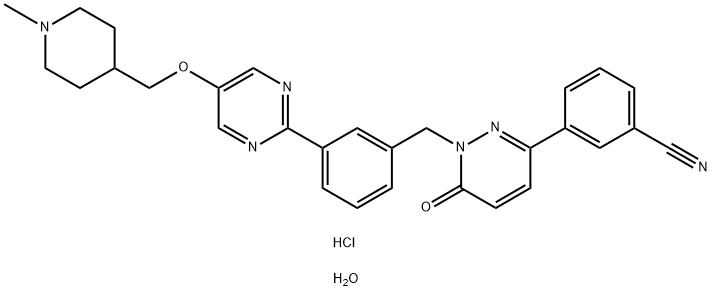 Tepotinib hydrochloride monohydrate
Tepotinib hydrochloride monohydrate
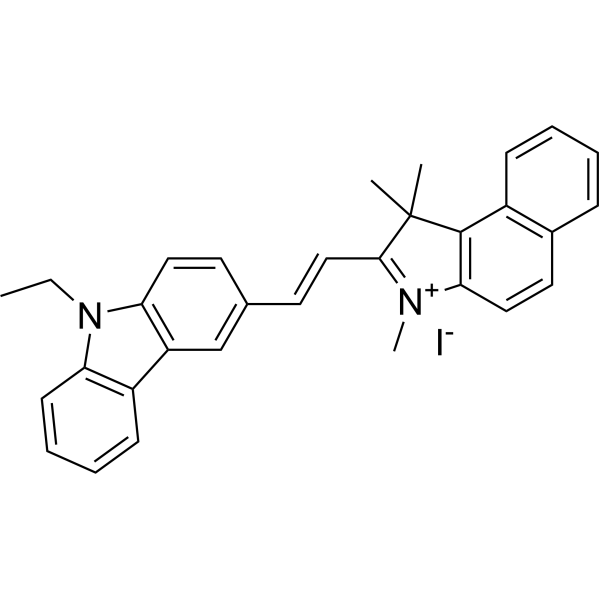 BKN-1
BKN-1
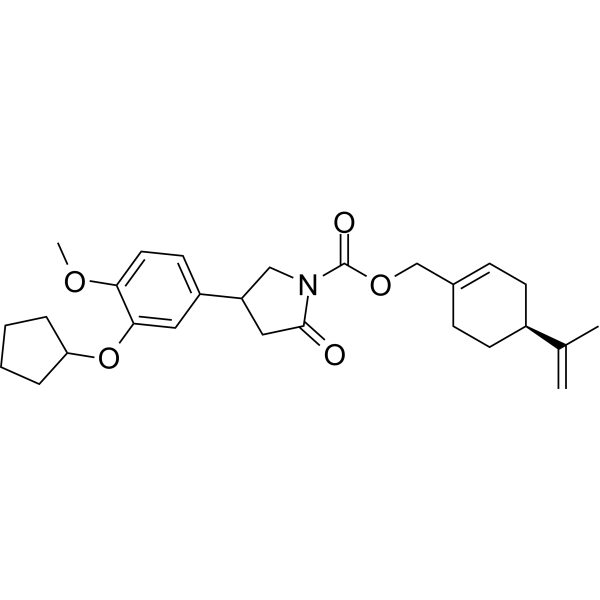 NEO214
NEO214
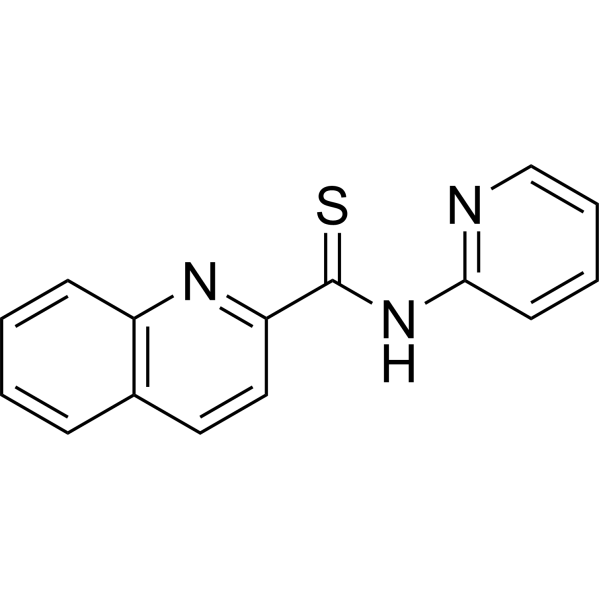 MJO445
MJO445
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
")
")
