| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
DNA Methyltransferase 32 nM (IC50) DNMT1 1.5 nM (Kd) DNMT3A 92 nM (IC50) DNMT3B 1000 nM (IC50) G9a 16 nM (IC50)
|
|---|---|
| 体外研究 (In Vitro) |
对于 DNMT1,CM-579 的 Kd 为 1.5 nM。 DNMT3A 和 DNMT3B 同样受到 CM-579 的抑制,IC50 值分别为 92 nM 和 1000 nM[1]。
|
| 体内研究 (In Vivo) |
CM-272 (CM-579 类似物)在体内具有抗白血病作用。[1]
将all来源的CEMO细胞(10 × 106)静脉注射于免疫缺陷的Rag2−/−γ - c−/−小鼠,每天给予2.5 mg kg−1 CM-272,注射后3天开始,持续28天。对照动物按相同方案给予生理盐水。与对照动物相比,CM-272治疗诱导小鼠的总生存期(OS)有统计学意义的增加(中位OS;92±5.7天vs 55±10.5天;P=0.0009)(图4a)。测定全肝提取物中H3K9me2和5mC的水平。通过流式细胞术(FACS)分析肝脏匀浆中的肿瘤浸润情况,发现60-80%的人细胞(hCD45+)浸润。治疗1周后,从动物身上获得的白血病细胞中,这两种标记都降低了(补充图10f)。治疗后的动物没有明显的体重减轻(补充图10g)。我们在como -1细胞的第二次体内复制中获得了类似的结果(补充图11a)。为了分析CM-272在体内的剂量依赖性功效,我们重复了同样的研究,给药1 mg kg - 1 CM-272(补充图12)。我们没有观察到动物体重的差异(补充图12a),也没有观察到1 mg kg - 1或2.5 mg kg - 1 CM-272与对照组之间的血液学参数的显著变化(补充图12b)。正如预期的那样,在2.5 mg kg - 1 CM-272处理的小鼠组中,CM-272血浆浓度更高(补充图12c)。然而,与2.5 mg kg - 1 CM-272不同,1 mg kg - 1 CM-272不能延长小鼠的存活时间(补充图12d)。这些结果表明CM-272的有效性是剂量依赖性的,2.5 mg kg - 1剂量的CM-272足以显示出积极的抗肿瘤功效。在第二种异种模型中,将10 × 106个aml来源的MV4-11细胞静脉注射Rag2 - / - γ - c - / -小鼠,14天后的动物用2.5 mg kg -1 CM-272治疗28天。与ALL细胞一样,CM-272治疗延长了小鼠的生存期(治疗组和未治疗组的中位生存期分别为78±12天和57±0.9天;P=0.0005)(图4b)。我们在MV4-11细胞的第二次体内复制中获得了类似的结果(补充图11b),没有任何毒性迹象(补充图10h)。最后,将OCI-Ly10活化的b细胞DLBCL细胞系2.5 × 106个细胞同样静脉注射到Rag2−/−γc−/−小鼠体内。与对照组相比,在8周内用相同剂量的CM-272治疗也延长了治疗小鼠的生存期(中位生存期;59±8天vs 49±6天;P=0.010)(图4c)。我们在OCI-Ly10细胞的第二次体内复制中获得了类似的结果(补充图11c),没有任何毒性迹象(补充图10i)。虽然对淋巴瘤细胞的影响在统计学上是显著的,但其效果不如AML和ALL细胞那么强。这些结果表明,CM-272通过抑制G9a/GLP和dnmt的甲基转移酶活性,在体内对不同类型的血液系统恶性肿瘤具有有效的抗肿瘤活性。除了上述信息外,与其他sam依赖的表观遗传酶相比,最小的混杂性(补充表4a和4b),进一步对癌症中其他药物靶点的脱靶选择性分析(一组97种激酶,补充表12-14)证实了G9a(和GLP)和dnmt是CM-272的主要靶点。 |
| 酶活实验 |
G9a和DNMT1酶活性测定[1]
采用时间分辨荧光能量转移法(TR-FRET)测定G9a和DNMT1活性。对于G9a,在G9a酶促反应后,将生物素化组蛋白单甲基H3K9肽与crypate标记的抗二甲基组蛋白H3K9抗体和链亲和素XL665孵育,观察TR-FRET。对于DNMT1,将Lumi4-Tb(供体)标记的s -腺苷型同型半胱氨酸特异性抗体与d2标记的s -腺苷型同型半胱氨酸(受体)一起孵育,使用EPIgeneous methyltransferase assay,观察到TR-FRET。详情请参阅补充资料。 针对G9a、DNMT1和GLP的放射性配体结合试验由Reaction Biology Corporation (http://www.reactionbiology.com)进行。 表观遗传选择性面板[1] 选择性的cm - 272和CM-579 对37表观遗传酶目标包括Bromodomain-containing酶(ATAD2A、ATAD2B BAZ2B, BRD1, BRD2(1型+ BD2), BRD4(1型+ BD2), BRDT(1型),CREBBP, TRIM24, TAF1),组蛋白甲基转移酶(EZH1, EZH2, GLP、PRMT1 PRMT3, PRMT4, PRMT5, PRMT6, PRMT8, SETD2, SET7/9, SUV39H1, SUV39H2和MLL-WARD), DNA甲基转移酶(种能阻碍DNMT3B DNMT3A和)和组蛋白demethylase (JMJD2A、JMJD2B JMJD2C, JMJD2D, JMJD2E, JMJD3, JMJD1A, LSD1, Jarid1A,Jarid1B和Jarid1C)由BPS Bioscience (http://www.bpsbioscience.com/index.ph)进行。 HDAC1和HDAC6酶活性测定[1] HDAC1和HDAC6酶的活性是用特定的荧光标记的底物来测定的,底物含有乙酰化的赖氨酸侧链,经过hdac去乙酰化。详情请参阅补充资料。 激酶选择性分析[1] 在DiscoverRx (http://www.discoverx.com/home)上使用KINOMEscan筛选平台,在10 μM的测试浓度下,对分布在kinome中的97个选定的激酶(其中90个是非突变激酶)进行CM-272的选择性分析。 直接结合分析[1] 采用微尺度热泳术(MST)定量CM-579 与DNMT1(全长)之间的生物分子相互作用。MST分析使用Monolith NT.115仪器进行。 ADME分析[1]< br > 以下ADME研究:CYP对人肝微粒体5种细胞色素p450 (1A2, 2C9, 2C19, 2D6和3A4, 10 μM)的抑制作用,血浆蛋白结合,动力学溶解度,Pampa渗透性和人和小鼠肝微粒体稳定性的研究由无锡公司(http://www.wuxi.com/)完成。 hERG阻断试验[1] 使用PredictorTM hERG荧光偏振商业检测试剂盒测定化合物对hERG钾通道的影响。 |
| 细胞实验 |
细胞增殖测定[1]
细胞类型: CEMO-1、MV4-11 和 OCI-Ly10 细胞系 测试浓度: 125 nM、250 nM , 500 nM(CEMO-1 细胞); 135 nM、270 nM、540 nM(MV4-11 细胞); 100 nM、400 nM、1000 nM(OCI-Ly10 细胞) 孵育时间:12 小时、24 小时、48 小时和 72 小时 实验结果:以剂量和时间依赖性方式抑制细胞增殖。 细胞周期分析 [1] 细胞类型: CEMO-1、MV4-11 和 OCI-Ly10 细胞系 测试浓度: > 125 nM、250 nM、500 nM(CEMO-1 细胞); 135 nM、270 nM、540 nM(MV4-11 细胞); 100 nM、400 nM、1000 nM(OCI-Ly10 细胞) 孵育时间: 24 小时 实验结果: 阻断细胞周期进程。 细胞凋亡分析[1] 细胞类型: CEMO-1、MV4-11 和 OCI-Ly10 细胞系 测试浓度: 125 nM、250 nM、500 nM(CEMO-1 细胞); 135 nM、270 nM、540 nM(MV4-11 细胞); 100 nM、400 nM、1000 nM(OCI-Ly10 细胞) 孵育时间:12 小时、24 小时、48 小时和 72 小时 实验结果:以剂量和时间依赖性方式诱导 ALL、AML 和 DLBCL 细胞系凋亡。 |
| 动物实验 |
Animal/Disease Models: Female BALB/Ca-Rag2−/−γc−/− mice (6–8weeks old) with CEMO-1 cells[1]
Doses: 2.5 mg/kg Route of Administration: intravenous (iv) injection; daily; for 28 days Experimental Results: Induced a statistically significant increase in overall survival (OS) in mice. PK study of CM-272 and CM-579 in plasma samples [1] CM-272 and CM-579 were measured in plasma samples using a Xevo-TQ MS triple quadropole mass spectrometer with an electrospray ionization (ESI) source and an Acquity UPLC. CM-272 and CM-579 solutions were prepared by dissolving the solid in saline. A drug dosage of 1 mg kg−1 or 2.5 mg kg−1 (CM-272) or 1 mg kg−1 (CM-579 ) was administered as a single intravenous injection. Blood was collected at predetermined times over 24 h post injection (0.25, 2, 4, 6, 8 and 24 h for CM-272) and (0.25, 1, 2, 4 and 8 h for CM-579). Chromatographic separation was performed by gradient elution at 0.6 ml min−1 using an Acquity UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm particle size). The PK parameters were obtained by fitting the blood concentration-time data to a non-compartmental model with the WinNonlin software. Details are provided in Supplementary Information. In vivo experiments[1] The human ALL CEMO-1 (control group with saline solution n=6; treated group with CM-272, n=6), AML MV4-11 (control group with saline solution n=8; treated group with CM-272 n=8) and DLBCL OCI-Ly10 (control group with saline solution n=6; treated group with CM-272 n=6) xenograft mice models were generated by i.v. injection of cells diluted in 100 μl of saline solution in the tail vein of a 6–8-week-old female BALB/cA−Rag2−/−γc−/− mice as described in Supplementary Information. CM-272 administration was detailed in Supplementary Information. Statistical results were calculated using the statistical software medcalc. CM-272 toxicity assay: haematological and liver parameters[1] After treating Rag2−/−γc−/− mice with daily i.v. 2.5 mg kg−1 of CM-272 during 4 weeks, followed by a 7 days washout period, haematological and liver parameters were measured as described in Supplementary Information. |
| 药代性质 (ADME/PK) |
Before performing PK studies, researchers identified the maximum tolerated dose (MTD) for CM-272, which it was at 2.5 mg kg−1 (intravenous, i.v.); however, for CM-579 we could not administer a dose higher than 1 mg kg−1 (i.v.). PK studies that researchers performed using the MTD dose (Supplementary Tables 9–11), showed clearance levels for CM-579 (5.7 l h−1 kg−1) and CM-272 (0.91 l h−1 kg−1).
|
| 毒性/毒理 (Toxicokinetics/TK) |
Before evaluating the in vivo efficacy of our dual inhibitors, researchers examined the therapeutic window achieved by these two molecules and their pharmacokinetic (PK) parameters. Thus, we studied toxicity of CM-272 and CM-579 using the non-tumoural hepatic cell line THLE-2 (LC50s were 1.78 and 1.30 μM) as well as peripheral blood mononuclear cells (PBMCs) obtained from healthy donors (LC50s were 1.90 and 7.39 μM) (Supplementary Table 2) and compared with the in vitro activity against tumour cell lines (Supplementary Table 3). Researchers detected that CM-272 and CM-579 showed an acceptable therapeutic window (around 1 log units). Before performing PK studies, we identified the maximum tolerated dose (MTD) for CM-272, which it was at 2.5 mg kg−1 (intravenous, i.v.); however, for CM-579 researchers could not administer a dose higher than 1 mg kg−1 (i.v.). PK studies that we performed using the MTD dose (Supplementary Tables 9–11), showed clearance levels for CM-579 (5.7 l h−1 kg−1) and CM-272 (0.91 l h−1 kg−1). [1]
|
| 参考文献 | |
| 其他信息 |
CM-272 is a member of the class of aminoquinolines that is is quinoline substituted by 5-methylfuran-2-yl, (1-methylpiperidin-4-yl)amino, methoxy, and 3-(pyrrolidin-1-yl)propoxy groups at positions 2, 4, 6 and 7, respectively. It is a dual G9a/DNA methyltransferases inhibitor with antitumor activity. It inhibits G9a, DNMT1, DNMT3A, DNMT3B and GLP (IC50 = 8 nM, 382 nM, 85 nM, 1200 nM and 2 nM, respectively). It has a role as an apoptosis inducer, a ferroptosis inducer, an antineoplastic agent, an EC 2.1.1.43 (enhancer of zeste homolog 2) inhibitor and an EC 2.1.1.37 [DNA (cytosine-5-)-methyltransferase] inhibitor. It is a N-alkylpyrrolidine, a member of furans, an aminoquinoline, an aromatic ether, a member of piperidines, a tertiary amino compound, a secondary amino compound and a diether.
The indisputable role of epigenetics in cancer and the fact that epigenetic alterations can be reversed have favoured development of epigenetic drugs. In this study, we design and synthesize potent novel, selective and reversible chemical probes that simultaneously inhibit the G9a and DNMTs methyltransferase activity. In vitro treatment of haematological neoplasia (acute myeloid leukaemia-AML, acute lymphoblastic leukaemia-ALL and diffuse large B-cell lymphoma-DLBCL) with the lead compound CM-272, inhibits cell proliferation and promotes apoptosis, inducing interferon-stimulated genes and immunogenic cell death. CM-272 significantly prolongs survival of AML, ALL and DLBCL xenogeneic models. Our results represent the discovery of first-in-class dual inhibitors of G9a/DNMTs and establish this chemical series as a promising therapeutic tool for unmet needs in haematological tumours.[1] The results of the transcriptomic analysis performed after treatment with CM-272 consistently suggest a tumour interferon type 1 response with expression of ISGs and induction of immunogenic cell death (ICD) in AML, ALL and DLBCL cells, pointing to a common mechanism of anti-tumour effect. Despite the fact that ICD had not been described as a mechanism of action for epigenetic drugs, these results might have been at least partially predicted, because recent studies have shown that expression of ISGs is epigenetically regulated by H3K9me2 (ref. 30), supporting the role of G9a inhibition in the activation of type I interferon responses and ICD. Thus, we speculate that the use of immunocompromised mice unable to develop anti-tumour immune responses may have underestimated the efficacy of CM-272 against tumour cells, prompting the evaluation of immune-competent models to explore the full potential therapeutic effects of our compounds33. On the basis of recent studies demonstrating that type I interferon responses contribute to the efficacy of chemotherapeutic agents32, the use of CM-272 in combination with such drugs and/or with immune modulators such as checkpoint inhibitors might also represent an attractive therapeutic strategy. In summary, CM-272 is a potent novel first-in-class dual reversible inhibitor of G9a (GLP) and DNMTs that prolongs survival in in vivo models of haematological malignancies by at least in part inducing immunogenic cell death. These compounds represent a novel approach for targeting cancer safely and efficiently, paving the way for treating a broad series of human tumours with poor prognosis.[1] |
| 分子式 |
C29H40N4O3
|
|---|---|
| 分子量 |
492.652907371521
|
| 精确质量 |
492.31
|
| 元素分析 |
C, 69.95; H, 7.61; N, 12.09; O, 10.35
|
| CAS号 |
1846570-40-8
|
| 相关CAS号 |
2448471-08-5 (CM-579 trihydrochloride)
|
| PubChem CID |
118613729
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
4.9
|
| tPSA |
63
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
10
|
| 重原子数目 |
36
|
| 分子复杂度/Complexity |
654
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
VMMRDFLDWHFNOX-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C29H40N4O3/c1-21-7-8-27(36-21)26-18-24(30-20-22-9-14-32(2)15-10-22)23-17-28(34-3)29(19-25(23)31-26)35-16-6-13-33-11-4-5-12-33/h7-8,17-19,22H,4-6,9-16,20H2,1-3H3,(H,30,31)
|
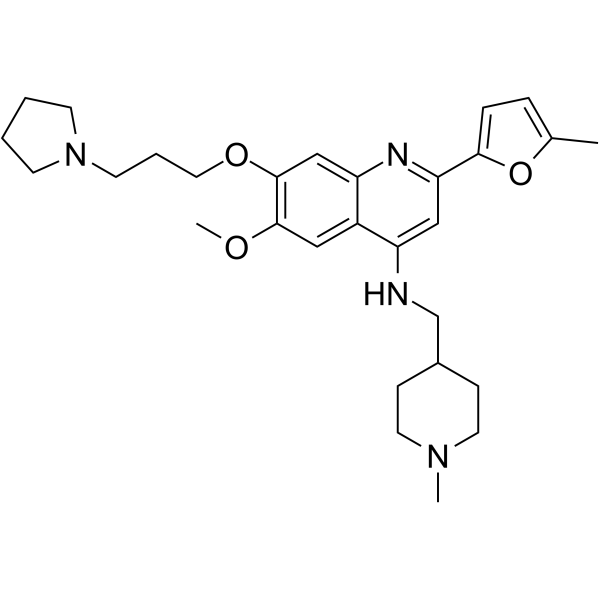
| 化学名 |
6-methoxy-2-(5-methylfuran-2-yl)-N-[(1-methylpiperidin-4-yl)methyl]-7-(3-pyrrolidin-1-ylpropoxy)quinolin-4-amine
|
| 别名 |
1846570-40-8; CM-579; CHEMBL4208004; CM579; CM 579; 6-Methoxy-2-(5-methylfuran-2-yl)-N-((1-methylpiperidin-4-yl)methyl)-7-(3-(pyrrolidin-1-yl)propoxy)quinolin-4-amine; 6-methoxy-2-(5-methylfuran-2-yl)-N-[(1-methylpiperidin-4-yl)methyl]-7-(3-pyrrolidin-1-ylpropoxy)quinolin-4-amine; 4-Quinolinamine, 6-methoxy-2-(5-methyl-2-furanyl)-N-[(1-methyl-4-piperidinyl)methyl]-7-[3-(1-pyrrolidinyl)propoxy]-; 6-methoxy-2-(5-methylfuran-2-yl)-N-[(1-methylpiperidin-4-yl)methyl]-7-[3-(pyrrolidin-1-yl)propoxy]quinolin-4-amine; SCHEMBL17379977;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0298 mL | 10.1492 mL | 20.2984 mL | |
| 5 mM | 0.4060 mL | 2.0298 mL | 4.0597 mL | |
| 10 mM | 0.2030 mL | 1.0149 mL | 2.0298 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































