
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
P2Y12 Receptor
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:氯吡格雷通过细胞色素 P450 (CYP) 酶转化为其活性代谢物。氯吡格雷 (1 μM) 还可抑制 RGM-1 细胞中 EGF 刺激的 EGF 受体、PERK 表达和细胞增殖 (P<0.05),并且对 EGF 受体过表达的 RGM-1 细胞中 EGF 刺激的细胞增殖的抑制作用要小得多细胞比 RGM-1 细胞减少(减少 22% 对 32%)。氯吡格雷增加血管数量,减少多形核计数,减少附着和骨丢失,还减少接受或未接受牙周修复的大鼠的破骨细胞数量。与盐水处理的大鼠相比,氯吡格雷降低了 CXCL4、CXCL12 和 PDGF 含量,但不影响 CXCL5。
|
| 体内研究 (In Vivo) |
氯吡格雷(2mg和10mg/kg/天)显着降低大鼠溃疡边缘溃疡诱导的胃上皮细胞增殖以及溃疡刺激的EGF受体和磷酸化细胞外信号调节激酶(PERK)的表达。氯吡格雷可改善充血性心力衰竭大鼠的内皮功能和一氧化氮生物利用度。氯吡格雷治疗的充血性心力衰竭 (CHF) 大鼠表现出增强 AKT 和 eNOS 的磷酸化。氯吡格雷/阿司匹林组合仅对兔耳横断引起的出血时间延长显示出累加型效应,因此表明环加氧酶和ADP效应的组合抑制提供了显着增强的抗血栓功效。
大鼠口服氯吡格雷或9a后氯吡格雷硫内酯的药代动力学参数 SD雄性大鼠口服24μmol kg-1的氯吡格雷或9a剂量。在给药前0小时和给药后0.25、0.5、1、2、4、6、8、24小时采集血液。SD雄性大鼠静脉注射8μmol kg-1的氯吡格雷硫内酯剂量,以确定氯吡格雷或9a转化为氯吡格雷硫内酯的转化率或生物利用度。在给药前0小时和给药后0.083、0.167、0.5、1、2、4、6、8、24小时采集血液。样品清理后,对血浆样品进行LC-MS/MS分析,以确定氯吡格雷硫内酯的血浆浓度(图5)。口服氯吡格雷后,血浆中氯吡格雷硫内酯的Cmax、Tmax、t1/2和AUC0-∞分别为6.93±3.36μg L-1、0.583±0.382 h、2.48±0.466 h和32.2±10.9μg·h L-1。口服9a后,血浆中氯吡格雷硫内酯的Cmax、Tmax、t1/2和AUC0-∞分别为67.2±42.3μg L-1、1.17±0.764 h、2.19±1.68 h和211±119μg·h L-1。上述数据证明,口服给药后,9a可以很容易地转化为氯吡格雷硫内酯,9a产生的氯吡格雷硫内酯的生物利用度比相同剂量的氯吡格雷高6倍。[1] |
| 酶活实验 |
在与肝微粒体的体外培养条件下,氯吡格雷可以通过硫内酯中间体(4a,Clopidogrel thiolactone/氯吡格雷硫内酯)代谢活化形成其活性代谢产物(AM,5)。形成的2-羟基四氢噻吩并吡啶是硫内酯4a的互变异构体,硫内酯4a是氯吡格雷第一次氧化活化产生的代谢中间体。氯吡格雷和9a的代谢活化途径在第一步通过2-羟基四氢噻吩并吡啶-硫内酯互变异构后汇合,并共享后续途径,包括NAPDH依赖的4a氧化环开放的第二步,最终导致活性代谢产物的形成[1]。
普拉格雷和氯吡格雷是抗血小板前药,通过硫内酯中间体转化为各自的活性代谢产物(例如Clopidogrel thiolactone/氯吡格雷硫内酯)。普拉格雷被酯酶快速水解为其硫内酯中间体,而氯吡格雷被细胞色素P450(CYP)异构体氧化为硫内酯。这两种硫内酯转化为活性代谢物是CYP介导的。本研究比较了普拉格雷和氯吡格雷硫内酯及其活性代谢产物在体内形成的效率。口服普拉格雷(1 mg kg(-1))和氯吡格雷(0.77 mg kg(-1))后,大鼠门静脉血浆中硫内酯中间体的血浆浓度-时间曲线下面积(AUC)分别为15.8+/-15.9 ng h ml(-1)和0.113+/-0.226 ng h ml。普拉格雷和氯吡格雷的活性代谢产物的相对生物利用度由活性代谢产物AUC(前药口服/活性代谢产物静脉注射)的比率计算得出,在大鼠中分别为25%和7%,在狗中分别为25%-10%。普拉格雷单次十二指肠内给药显示普拉格雷完全转化,导致大鼠门静脉血浆中硫内酯和普拉格雷的活性代谢产物浓度很高,这表明这些产物是在吸收过程中在肠道中产生的。总之,普拉格雷的硫内酯和活性代谢产物的体内形成程度大于氯吡格雷的硫内酯及其活性代谢产物。[2] |
| 动物实验 |
Sprague-Dawley male rats
8 μm/kg; orally clopidogrel (24 μm/kg) Intravenous injection; collected blood at 0 h (before dosing) and 0.083, 0.167, 0.5, 1, 2, 4, 6, 8, 24 h postdose. Pharmacokinetic Studies[1] Sprague–Dawley male rats (SPF grade) were used to determine oral bioavailability and PK parameters of the clopidogrel and 9a following oral and intravenous administration. In this study, Clopidogrel Thiolactone (4a) was measured by LC/MS/MS analysis to determine the PK parameters for both clopidogrel and 9a. PK parameters were calculated using a noncompartmental model. Solutions of clopidogrel and 9a in N,N-dimethylacetamide (DMA)/polyethylene glycol 400 (PEG 400) (v/v 5:95) at 1.26 and 1.14 mg/mL were prepared for oral dosing. A solution of clopidogrel thiolactone in DMA/PEG 400/saline (v:v:v 5:15:80) at 0.54 mg/mL was prepared for intravenous administration to determine the oral bioavailability of both clopidogrel and 9a to be converted to clopidogrel thiolactone. The solution formulations of clopidogrel thiolactone along with clopidogrel and 9a were administered separately by bolus injection into a tail vein and by oral gavage. Blood samples (about 0.20 mL) were collected from the retro-orbital plexus from three animals in each treatment group at 0 (predose), 0.083, 0.167. 0.5, 1, 2, 4, 6, 8, and 24 h for iv and 0 (predose), 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h for po. Calibration standards with clopidogrel thiolactone concentrations from 0.5 to 1000 ng/mL were prepared by serial dilution in pretreated naive rat plasma. A portion (25 μL) of each calibration standard and unknown study sample was mixed with 25 μL of acetonitrile containing the internal standard followed by addition of 200 μL of MTBE into each sample. All samples were vortex-mixed for 3 min. The mixture was then centrifuged at 15700g at 4 °C for 3 min. The supernatants containing the organic component for each sample were used for analysis. The lower limit of quantitation was 0.5 ng/mL. |
| 参考文献 |
|
| 其他信息 |
2-oxoclopidogrel is an organic molecular entity.
|
| 分子式 |
C16H16CLNO3S
|
|
|---|---|---|
| 分子量 |
337.82
|
|
| 精确质量 |
337.053
|
|
| 元素分析 |
C, 56.89; H, 4.77; Cl, 10.49; N, 4.15; O, 14.21; S, 9.49
|
|
| CAS号 |
1147350-75-1
|
|
| 相关CAS号 |
Clopidogrel; 113665-84-2; Clopidogrel hydrogen sulfate; 120202-66-6
|
|
| PubChem CID |
56848893
|
|
| 外观&性状 |
Brown to reddish brown solid powder
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 沸点 |
488.1±45.0 °C at 760 mmHg
|
|
| 闪点 |
249.0±28.7 °C
|
|
| 蒸汽压 |
0.0±1.2 mmHg at 25°C
|
|
| 折射率 |
1.640
|
|
| LogP |
2.96
|
|
| tPSA |
71.91
|
|
| 氢键供体(HBD)数目 |
0
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
22
|
|
| 分子复杂度/Complexity |
496
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
ClC1=C([H])C([H])=C([H])C([H])=C1[C@@]([H])(C(=O)OC([H])([H])[H])N1C([H])([H])C2=C([H])C(=O)SC2([H])C([H])([H])C1([H])[H]
|
|
| InChi Key |
JBSAZVIMJUOBNB-WUJWULDRSA-N
|
|
| InChi Code |
InChI=1S/C16H16ClNO3S/c1-21-16(20)15(11-4-2-3-5-12(11)17)18-7-6-13-10(9-18)8-14(19)22-13/h2-5,8,13,15H,6-7,9H2,1H3/t13?,15-/m0/s1
|
|
| 化学名 |
methyl (2S)-2-(2-chlorophenyl)-2-(2-oxo-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-5-yl)acetate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.03.00
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (7.40 mM) (饱和度未知) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80+,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9602 mL | 14.8008 mL | 29.6016 mL | |
| 5 mM | 0.5920 mL | 2.9602 mL | 5.9203 mL | |
| 10 mM | 0.2960 mL | 1.4801 mL | 2.9602 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
J Med Chem.2012 Apr 12;55(7):3342-52. |
|---|
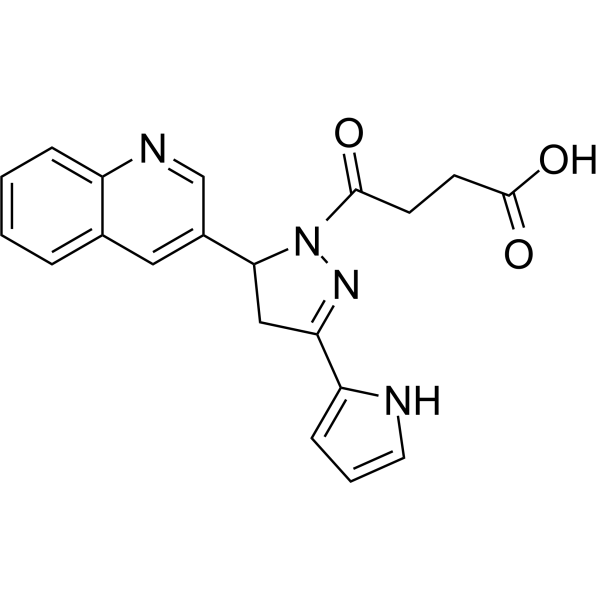 P2Y6R antagonist 1
P2Y6R antagonist 1
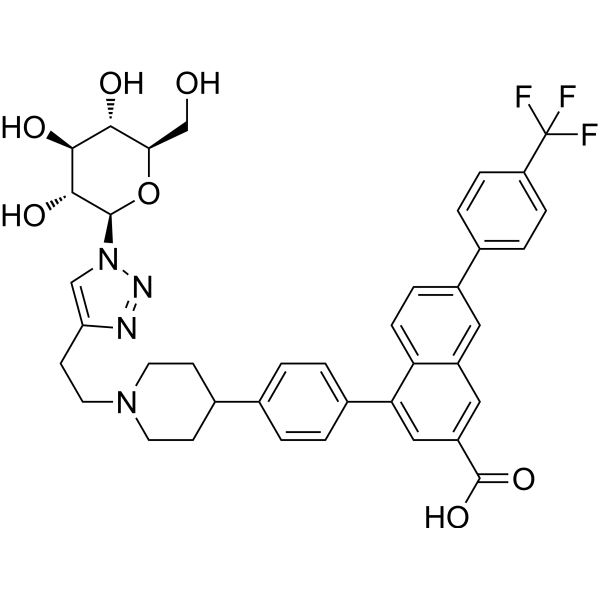 MRS4865
MRS4865
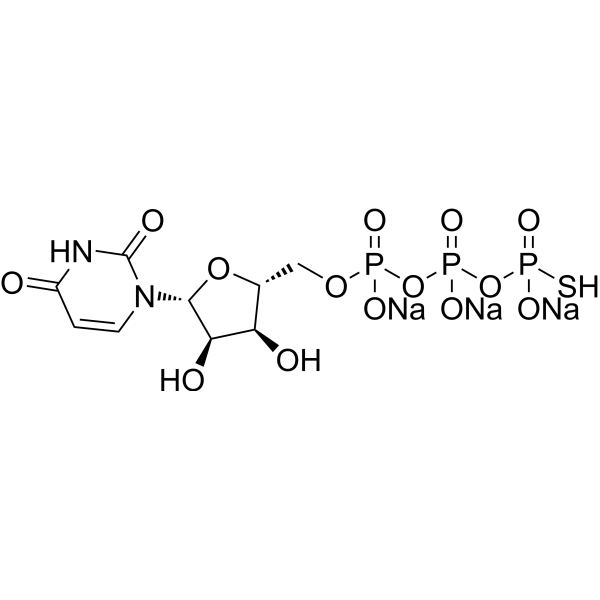 Uridine-5'-O-(3-thiotriphosphate) trisodium
Uridine-5'-O-(3-thiotriphosphate) trisodium
 MRS4833
MRS4833
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA

