| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| 50g |
|
||
| 100g |
|
||
| Other Sizes |
|

| 靶点 |
Endogenous Metabolite; nuclear receptors (FXR)
|
|---|---|
| 体外研究 (In Vitro) |
鹅去氧胆酸 (CDCA) 和脱氧胆酸 (DCA) 的 IC50 值分别为 22 mM 和 38 mM,均能阻断 11 β HSD2,诱导酒精旋转,并提高盐应激激素 (MR) 的调节活性 [1]。通过激活膜G蛋白偶联受体(TGR5),螯合去氧胆酸可以显着提高细胞周期蛋白D1蛋白和mRNA的表达,从而促进Ishikawa细胞的发育[2]。在培养的人肝母细胞瘤细胞系 Hep G2 中,鹅去氧胆酸 (CDCA) 可以降低 HMG-CoA 还原酶和 HMG-CoA 合酶的 mRNA 水平,并使 LDL 受体 mRNA 水平提高约四倍[3]。有两个级别的提升。布美他尼、BaCl2 和囊性纤维化跨膜电导调节剂 (CFTR) 激活 CFTRinh-172,可抑制鹅去氧胆酸产生的 Isc (≥67%)。腺苷酸环化酶镜像 MDL12330A 使鹅去氧胆酸刺激的 Isc 降低了 43%,但鹅去氧胆酸提高了细胞内 cAMP 浓度 [4]。 HepG2 细胞中的表达增加、磷酸化和核积累证明了鹅去氧胆酸治疗会激活 C/EBPβ。带有 C/EBP 响应元件 (pGL-1651) 的 1.65-kb GSTA2 启动子用于控制鹅去氧胆酸增强的荧光素酶基因。采用 AMPKα 显性失活突变体和化学替代的实验研究表明,鹅去氧胆酸疗法可激活 AMP 激活蛋白状态 (AMPK),从而导致细胞外信号调节因子 1/2 (ERK1/2) 的激活 [5]。
矿皮质激素受体(MR)的不适当激活导致肝硬化患者肾脏钠潴留和钾流失。最近的证据表明,这种MR激活至少部分是胆汁酸依赖的11 β -羟基类固醇脱氢酶2 (11 β HSD2)活性降低的结果,这种酶通过将皮质醇转化为可的松来防止皮质醇依赖的MR激活。在这里,我们研究了胆汁酸介导的MR激活的分子机制。12例胆道梗阻患者的尿胆汁酸分析显示,鹅去氧胆酸(CDCA)、胆酸(CA)和去氧胆酸(DCA)浓度均较高,平均浓度为50-80微米。虽然在瞬时表达的HEK-293细胞中,Chenodeoxycholic acid (CDCA)和DCA均在缺乏11 β HSD2和类固醇的情况下介导MR的核易位,但MR的转录活性并未受到刺激。相比之下,CDCA和DCA分别以22和38微米的IC(50)值抑制11 β HSD2,并引起皮质醇依赖的核易位和MR转录活性增加。LCA,最有效抑制11 β HSD2的胆汁酸,在胆汁沉积症患者中以非常低的浓度存在,而弱抑制剂CA不会引起MR激活。总之,这些发现表明,CDCA(在较小程度上DCA)通过抑制11 β HSD2介导皮质醇依赖性核易位和MR的转录激活,并至少在胆道梗阻患者中观察到钠潴存和钾排泄的部分原因。[1] 子宫内膜癌在西方发达国家发病率高,主要原因是高脂肪饮食和肥胖。膳食脂的加工是由胆汁酸触发的,胆汁酸是在肝脏合成并储存在胆囊中的两相洗涤剂。胆汁酸除了在膳食脂质吸收和胆固醇稳态中发挥众所周知的作用外,还可以作为具有全身内分泌功能的信号分子。在本研究中,我们研究了原发性胆汁鹅去氧胆酸/Chenodeoxycholic acid (CDCA)对人子宫内膜癌细胞系Ishikawa的生物学效应。低浓度的CDCA通过激活膜G蛋白偶联受体(TGR5)依赖通路,诱导Cyclin D1蛋白和mRNA表达显著增加,从而刺激石川细胞生长。EMSA和ChIP分析表明,cdca诱导的Cyclin D1表达需要转录因子CREB在Cyclin D1基因近端启动子内环amp响应元件基序上的增强募集。我们的研究结果提出了一种新的分子机制,解释了高脂肪饮食和肥胖对子宫内膜癌生长和进展的潜在贡献,为预防这种与肥胖相关的女性癌症风险的策略提供了理论依据。[2] 在培养的人肝母细胞瘤细胞系Hep G2中,<强>鹅脱氧胆酸(CDCA)诱导LDL受体mRNA水平约为4倍,HMG-CoA还原酶和HMG-CoA合成酶mRNA水平为2倍。甲戊酸激酶、法尼酯焦磷酸合成酶和角鲨烯合成酶mRNA水平变化不显著。甾醇敏感基因的诱导模式与SREBP降解抑制剂N-acetyl-leucyl-leucyl-norleucinal (ALLN)的诱导模式相似,提示CDCA可能会增加成熟SREBP。CDCA可以抑制25-羟基胆固醇介导的SREBP失活,但不影响SREBP的mRNA水平。这些结果表明,CDCA可以通过一种新机制影响固醇代谢,该机制涉及抑制氧甾醇介导的SREBP失活。[3] 高水平的Chenodeoxycholic acid (CDCA)/去氧胆酸刺激哺乳动物结肠上皮中Cl(-)的分泌。虽然不同的第二信使参与了这一作用,但具体的信号通路尚未完全描述。在人结肠癌T84细胞中,我们通过测量I(-)外排和短路电流(Isc),阐明了这个级联评估Cl(-)转运。CDCA (500 μM)迅速增加I(-)外排,我们通过Isc证实,当它添加到基底外侧比添加到根尖表面时,会引起更大的响应。然而,细胞因子的预孵育使单层对顶端添加的反应性提高了55%。制霉菌素渗透性研究表明,CDCA刺激产生电的顶端Cl(-)电流,而不是基底侧K(+)电流。此外,cdca诱导的Isc被布美他尼、BaCl2和囊性纤维化跨膜传导调节剂(CFTR)抑制剂CFTRinh-172抑制(≥67%)。腺苷酸环化酶抑制剂MDL12330A刺激的Isc降低43%,CDCA增加细胞内cAMP浓度。蛋白激酶A抑制剂H89和微管破坏剂诺可达唑分别导致cdca刺激的Isc减少94%和47%。用CFTR抗体进行免疫沉淀,然后用Pan-phospho和CFTR抗体进行顺序免疫印迹,结果显示CDCA使CFTR磷酸化增加了大约两倍。对CDCA反应的快速和侧边特异性暗示了一个膜介导的过程。虽然CDCA的作用不被毒蕈碱受体拮抗剂阿托品阻断,但T84细胞具有胆汁酸G蛋白偶联受体TGR5的转录物和蛋白。这些结果首次证明CDCA通过涉及微管的cAMP-PKA通路激活CFTR,并暗示这是通过基底外侧膜受体发生的。[4] Farnesoid X受体(FXR)调节氧化还原稳态并引起细胞保护作用。CCAAT/增强子结合蛋白-β (C/EBPβ)参与调节肝细胞特异性基因的表达,参与肝细胞保护和肝脏再生。鉴于FXR在外源代谢和肝细胞存活中的作用,本研究探讨了FXR激活C/EBPβ诱导解毒酶及其调控途径的潜力。鹅脱氧胆酸/Chenodeoxycholic acid (CDCA)是胆汁酸的主要成分,可激活FXR。在HepG2细胞中,Chenodeoxycholic acid (CDCA)处理激活了C/EBPβ,其磷酸化,核积累和表达增加。合成FXR配体3-(2,6-二氯苯基)-4-(3'-羧基-2-氯二苯基-4-基-)氧甲基-5-异丙基异恶唑(GW4064)也具有类似的效果。此外,CDCA增强了含有-1.65 kb GSTA2启动子的荧光素酶基因转录,该启动子含有C/EBP应答元件(pGL-1651)。此外,CDCA处理激活了amp活化蛋白激酶(AMPK),从而导致细胞外信号调节激酶1/2 (ERK1/2)活化,使用AMPKα显性阴性突变体和化学抑制剂的实验结果证明了这一点。ERK1/2的激活负责激活C/EBPβ的磷酸化。FXR敲低降低了CDCA激活AMPK和ERK1/2以及磷酸化C/EBPβ的能力。FXR的表达促进了AMPKα、ERK1/2和C/EBPβ的磷酸化,证实了CDCA引起的C/EBPβ磷酸化是由FXR激活AMPK和ERK1/2引起的。在小鼠中,CDCA处理通过诱导肝脏解毒酶激活C/EBPβ。我们的研究结果表明,CDCA通过ampk依赖性ERK1/2通路激活C/EBPβ,从而诱导抗氧化和外源代谢酶。 |
| 体内研究 (In Vivo) |
用鹅去氧胆酸/Chenodeoxycholic acid (CDCA)治疗的大鼠血压升高,用Chenodeoxycholic acid (CDCA)治疗的去肾上腺大鼠肾脏钠潴留和尿钾排泄增强。11ß-羟基类固醇脱氢酶(11β-HSD)将活性糖皮质激素代谢为其无活性的11-脱氢产物,并保护肾矿化皮质激素受体免受高循环水平内源性糖皮质激素的影响。11ß-HSD被认为不仅在控制肾钠潴留方面很重要,而且在控制血压方面也很重要。我们之前已经证明,1α-和11ß-羟孕酮(11α-和11ß-OHP)是(I)体外11ß-HSD(异构体1和2)活性的有效抑制剂,(ii)体内皮质酮(B)能够赋予矿化皮质激素(MC)活性,(iii)长期输注于Sprague-Dawley (SD)大鼠时可致高血压。此外,我们还发现3α, 5b -四氢孕酮(3α,5B-THP)和鹅去氧胆酸(CDCA)是11ß-HSD1活性的有效抑制剂,而不是11ß-HSD2活性的抑制剂,然而,这些物质仍然能够在肾上腺切除大鼠中赋予B MC活性。为了评估3α、5B-THP和CDCA可能的血压调节作用,我们现在将这些物质连续注入完整的SD大鼠体内14天。3α、5B-THP和CDCA均在7天内引起血压显著升高,这种影响在14天的输注中持续存在。上述结果表明,3α、5B-THP和CDCA在大鼠中均具有致高血压作用,内源性孕酮代谢物和CDCA对11ß-HSD2或11ß-HSD1活性的抑制可能参与了高血压的病理过程。https://www.tandfonline.com/doi/abs/10.1080/07435809609043778
|
| 酶活实验 |
胆汁酸抑制11βHSD2活性的测定[1]
11βHSD2酶活性在细胞裂解物中测量,如所述。简单地说,转染的HEK-293细胞,在炭处理过的Dulbecco改良Eagle培养基中培养24小时,用Hanks液洗涤一次,并在含有100 mm NaCl, 1 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 250 mm蔗糖,20 mm Tris-HCl, pH 7.4的缓冲液中重悬。细胞在液氮中冷冻裂解。在最终体积为20 μl,含有适当浓度的胆汁酸,30 nCi的[3H]皮质醇和未标记的皮质醇至最终浓度为50 nm时,测量脱氢酶活性。将细胞裂解液与反应混合物混合,开始反应。或者,根据前面描述的程序,从转染的HEK-293细胞制备内质网微粒体,并与含有不同浓度的皮质醇和Chenodeoxycholic acid (CDCA)的反应混合物孵育。培养20分钟,用薄层色谱(TLC)测定皮质醇向可的松的转化。由于TLC方法在低转化率时不准确,而在转化率高于60-70%时,最终产物对11βHSD2的抑制作用较大,因此仅考虑10 - 60%之间的转化率进行计算。采用曲线拟合程序评估抑菌常数IC50。结果以平均值±标准差表示,并由至少四个独立的测量组成 为了确定11βHSD2是否代谢Chenodeoxycholic acid (CDCA),采用气相色谱-质谱法分析瞬时表达11βHSD2的HEK-293细胞培养的上清。1 ml细胞培养上清中加入4 μg的23-去氧胆酸作为回收标准液,在Sep-Pak C18色谱柱上提取。提取液中加入4 μg的5β-胆甾-3β-醇作为衍生化气相色谱标准品,衍生得到甲基lester三甲基硅醚。在Lipidx-5000色谱柱上通过凝胶过滤除去多余的硅化剂。样品采用气相色谱-质谱法分析,使用惠普6890气相色谱仪,配备质量选择检测器5973,通过选择离子监测(SIM,针对5种不同的胆汁酸编程),并在扫描模式下检测由Chenodeoxycholic acid (CDCA)形成的任何潜在的新代谢物。 |
| 细胞实验 |
碘化流出。[4]
碘化物外排研究如我们前面所述,基于Venglarik等人的原始方法,经Chappe等人修改。简单地说,T84细胞在六孔板中生长。每孔接种100万个细胞,4-5天使细胞达到90%的合度,然后在室温下黑暗中用碘化负载缓冲液(含mM: 136 NaI, 3 KNO3, 2 Ca(NO3)2, 11葡萄糖和20 HEPES pH 7.4)孵育1小时。然后用无碘的外排缓冲液冲洗细胞三次(与碘离子负载缓冲液相同,只是NaNO3取代了NaI)。单个井暴露于DMSO、Chenodeoxycholic acid (CDCA) (500 μM)、TCDC (500 μM)或由100 μM 8-Br-cAMP + 10 μM forskolin + 100 μM IBMX组成的cAMP混合物中。然后将流出缓冲液(1ml)加入培养皿中;2分钟后,取出缓冲液并保存,每孔加入1 ml新鲜流出缓冲液。因此,在实验期间每隔2分钟收集一次样品。采用碘离子敏感电极和pH/mV计测定样品中的碘离子浓度。根据前面描述的标准曲线计算样品的碘化物浓度,并将其描述为每隔2分钟碘化物流出率(nmol/min)。 免疫沉淀和免疫印迹分析。[4] 参照Sakesena等的方法进行免疫沉淀和免疫印迹分析。这些细胞的生长和处理方案类似于ususing chamber实验。简单地说,将T84细胞以1.5 × 106个细胞的密度接种于六孔Transwell组织培养插入物中。经EVOM2伏安计和STX2电极测定,当TER值≥950 Ω·cm2(~ 9-14天)时,用DMSO(0.1%)、Chenodeoxycholic acid (CDCA) (500 μM)或forskolin (10 μM)对细胞进行基底侧处理20 min。PBS洗涤细胞3次,将膜切割并浸入裂解缓冲液(20 mM Tris·HCl pH 7.5、150 mM NaCl、1% Triton X-100、1 mM EDTA、1 mM EGTA、蛋白酶抑制剂混合物)中。和磷酸酶抑制剂混合物2)。4个孔的细胞合并为一个样本。细胞在冰上超声处理(25 s)。匀浆在4°C下离心(1000 g, 10 min),使细胞核和未破裂的细胞成球。将含5 mg蛋白的上清液与3 μg单克隆抗人CFTR cooh末端抗体在摇床上4℃孵育过夜。孵育后,用蛋白A/G +琼脂糖免疫沉淀试剂沉淀免疫复合物。 |
| 动物实验 |
3α, 5β-THP and bile acids were also tested to determine whether, like GA, they could confer mineralocorticoid actions upon corticosterone (B). In adrenalectomized rats pretreated with Chenodeoxycholic acid (CDCA) or 3α,5β-THP, B caused a significant antinatriuresis; the effect of B plus CDCA was blocked by the antimeneralocorticoid, RU 28318. Thus we report on two structurally similar endogenous substance, 3α, 5β-THP and CDCA, which inhibit both 11β-OHSD and 5β-R activity, and which can confer mineralocorticoid upon the glucococorticoid, B. (Steroids 59: 352–356, 1994), https://www.sciencedirect.com/science/article/abs/pii/0039128X94900019
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Chenodiol is well absorbed from the small intestine. About 80% of its bacterial metabolite lithocholate is excreted in the feces. Metabolism / Metabolites Chenodiol is well absorbed from the small intestine and taken up by the liver where it is converted to its taurine and glycine conjugates and secreted in bile. At steady-state, an amount of chenodiol near the daily dose escapes to the colon and is converted by bacterial action to lithocholic acid. About 80% of the lithocholate is excreted in the feces; the remainder is absorbed and converted in the liver to its poorly absorbed sulfolithocholyl conjugates. During chenodiol therapy there is only a minor increase in biliary lithocholate, while fecal bile acids are increased three- to fourfold. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In multiple clinical trials of chenodiol therapy for dissolution of gallstones, serum aminotransferase elevations occurred in up to 30% of patients. The elevations generally arose within 2 months of starting therapy and were typically mild, transient and not accompanied by symptoms or jaundice. Liver biopsies done during chenodiol therapy generally showed mild, nonspecific changes. Clinically apparent liver injury with jaundice was not reported. The liver enzyme elevations were generally dose related and usually did not recur on restarting chenodiol at lower doses. While the serum enzyme abnormalities that occurred on chenodiol therapy generated considerable concern, they appeared to be relatively benign. Since the approval of chenodiol and its more widespread use, at least four instances of liver injury with jaundice have been reported to the sponsor, but the clinical features and outcomes of these cases have not been published. Nevertheless, the product label for chenodiol includes a boxed warning about hepatotoxicity although it does not provide advice on the frequency or how to respond to abnormalities. Thus, the reliability of reports of clinically apparent liver injury with chenodiol therapy remains unclear. Once ursodiol was found to be equally as effective as chenodiol, even at lower doses, and was rarely associated with serum enzyme elevations, it rapidly replaced chenodiol as medical therapy for gallstones. Likelihood score: E* (Suspected but unproven cause of clinically apparent liver injury). 10133 rat LD50 oral 4 gm/kg BEHAVIORAL: CHANGES IN MOTOR ACTIVITY (SPECIFIC ASSAY); LUNGS, THORAX, OR RESPIRATION: DYSPNEA; GASTROINTESTINAL: HYPERMOTILITY, DIARRHEA Oyo Yakuri. Pharmacometrics., 15(915), 1978 10133 rat LD50 intraperitoneal 105 mg/kg BEHAVIORAL: SOMNOLENCE (GENERAL DEPRESSED ACTIVITY); GASTROINTESTINAL: OTHER CHANGES; SKIN AND APPENDAGES (SKIN): HAIR: OTHER Oyo Yakuri. Pharmacometrics., 15(915), 1978 10133 rat LD50 subcutaneous >4 gm/kg BLOOD: CHANGES IN SPLEEN; SKIN AND APPENDAGES (SKIN): HAIR: OTHER Kiso to Rinsho. Clinical Report., 11(2499), 1977 10133 rat LD50 intravenous 106 mg/kg BEHAVIORAL: SOMNOLENCE (GENERAL DEPRESSED ACTIVITY); BEHAVIORAL: CONVULSIONS OR EFFECT ON SEIZURE THRESHOLD; BEHAVIORAL: ATAXIA Oyo Yakuri. Pharmacometrics., 15(915), 1978 10133 rat LD50 intramuscular >500 mg/kg Drugs in Japan, -(397), 1990 |
| 参考文献 |
|
| 其他信息 |
Chenodiol can cause developmental toxicity according to state or federal government labeling requirements.
Chenodeoxycholic acid is a dihydroxy-5beta-cholanic acid that is (5beta)-cholan-24-oic acid substituted by hydroxy groups at positions 3 and 7 respectively. It has a role as a human metabolite and a mouse metabolite. It is a bile acid, a dihydroxy-5beta-cholanic acid and a C24-steroid. It is a conjugate acid of a chenodeoxycholate. Chenodeoxycholic acid (or Chenodiol) is an epimer of ursodeoxycholic acid (DB01586). Chenodeoxycholic acid is a bile acid naturally found in the body. It works by dissolving the cholesterol that makes gallstones and inhibiting production of cholesterol in the liver and absorption in the intestines, which helps to decrease the formation of gallstones. It can also reduce the amount of other bile acids that can be harmful to liver cells when levels are elevated. Chenodeoxycholic acid (chenodiol) is a primary bile acid, synthesized in the liver and present in high concentrations in bile that is used therapeutically to dissolve cholesterol gallstones. Chronic therapy is associated with transient elevations in serum aminotransferase levels in up to 30% of patients, but chenodiol has been linked to only rare instances of clinically apparent liver injury with jaundice. Chenodeoxycholic acid has been reported in Homo sapiens and Ganoderma lucidum with data available. A bile acid, usually conjugated with either glycine or taurine. It acts as a detergent to solubilize fats for intestinal absorption and is reabsorbed by the small intestine. It is used as cholagogue, a choleretic laxative, and to prevent or dissolve gallstones. See also: Sodium Chenodeoxycholate (is active moiety of). Drug Indication Chenodiol is indicated for patients with radiolucent stones in well-opacifying gallbladders, in whom selective surgery would be undertaken except for the presence of increased surgical risk due to systemic disease or age. Chenodiol will not dissolve calcified (radiopaque) or radiolucent bile pigment stones. FDA Label Chenodeoxycholic acid is indicated for the treatment of inborn errors of primary bile acid synthesis due to sterol 27 hydroxylase deficiency (presenting as cerebrotendinous xanthomatosis (CTX)) in infants, children and adolescents aged 1 month to 18 years and adults. Mechanism of Action Chenodiol suppresses hepatic synthesis of both cholesterol and cholic acid, gradually replacing the latter and its metabolite, deoxycholic acid in an expanded bile acid pool. These actions contribute to biliary cholesterol desaturation and gradual dissolution of radiolucent cholesterol gallstones in the presence of a gall-bladder visualized by oral cholecystography. Bile acids may also bind the the bile acid receptor (FXR) which regulates the synthesis and transport of bile acids. |
| 分子式 |
C24H40O4
|
|---|---|
| 分子量 |
392.58
|
| 精确质量 |
392.292
|
| 元素分析 |
C, 73.43; H, 10.27; O, 16.30
|
| CAS号 |
474-25-9
|
| 相关CAS号 |
Chenodeoxycholic Acid-d4;99102-69-9;Chenodeoxycholic acid-13C;52918-92-0;Chenodeoxycholic Acid-d9;Chenodeoxycholic acid-d5;52840-12-7
|
| PubChem CID |
10133
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
547.1±25.0 °C at 760 mmHg
|
| 熔点 |
165-167 °C(lit.)
|
| 闪点 |
298.8±19.7 °C
|
| 蒸汽压 |
0.0±3.3 mmHg at 25°C
|
| 折射率 |
1.543
|
| LogP |
4.66
|
| tPSA |
77.76
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
28
|
| 分子复杂度/Complexity |
605
|
| 定义原子立体中心数目 |
10
|
| SMILES |
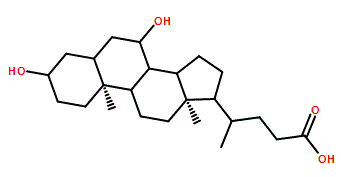
C[C@H](CCC(=O)O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2[C@@H](C[C@H]4[C@@]3(CC[C@H](C4)O)C)O)C
|
| InChi Key |
RUDATBOHQWOJDD-BSWAIDMHSA-N
|
| InChi Code |
InChI=1S/C24H40O4/c1-14(4-7-21(27)28)17-5-6-18-22-19(9-11-24(17,18)3)23(2)10-8-16(25)12-15(23)13-20(22)26/h14-20,22,25-26H,4-13H2,1-3H3,(H,27,28)/t14-,15+,16-,17-,18+,19+,20-,22+,23+,24-/m1/s1
|
| 化学名 |
(4R)-4-[(3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanoic acid
|
| 别名 |
Anthropodesoxycholic acid; Anthropodeoxycholic acid; chenodeoxycholic acid; Chenodiol; 474-25-9; Chenix; Chenic acid; Cdca; Chenodeoxycholate; Chenodeoxycholic Acid
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ≥ 50 mg/mL (~127.37 mM)
0.1 M NaOH : ~50 mg/mL (~127.37 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.5 mg/mL (6.37 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.37 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.37 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 20 mg/mL (50.95 mM) (饱和度未知) in 20% HP-β-CD in Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5473 mL | 12.7363 mL | 25.4725 mL | |
| 5 mM | 0.5095 mL | 2.5473 mL | 5.0945 mL | |
| 10 mM | 0.2547 mL | 1.2736 mL | 2.5473 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































