| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
Histamine receptor
|
|---|---|
| 体外研究 (In Vitro) |
高剂量的西替利嗪 (>5 μM) 可抑制经 IL-1β 处理的 A549 细胞释放 GM-CSF 和 IL-8。西替利嗪除了作为组胺 H1 受体拮抗剂外,还具有抗炎特性[2]。
|
| 体内研究 (In Vivo) |
当小鼠接触豚草花粉并进行免疫时,西替利嗪(20 mg/kg,小鼠)可抑制 IL-8 和 MIF 的产生,从而减轻炎症[3]。
|
| 酶活实验 |
最近的研究表明,除了H(1)受体拮抗作用外,几种第二代抗组胺药还可以调节各种炎症反应。抗组胺药西替利嗪是左西替利辛和右西替利津的外消旋混合物。本研究的目的是研究这两种抗组胺药(西替利嗪和左西替利津)对A549人气道上皮细胞粒细胞-巨噬细胞集落刺激因子(GM-CSF)和白细胞介素(IL)-8分泌的影响。将A549细胞分别与西替利嗪(0.1、1、2.5、5和10μM)或左西替利津(0.1、2、5、和10μm)预孵育16小时,然后用IL-1β刺激8小时。采用酶联免疫吸附试验(ELISA)测定培养上清液中GM-CSF和IL-8的水平。我们的数据显示,西替利嗪(5和10微克)和左西替利津(2.5、5和10微米)显著抑制了IL-1β刺激的A549细胞的GM-CSF分泌(p<0.05)。在刺激A549后,西替利嗪(10μM)和左西替利津(5和10μm)显著抑制IL-8的分泌。2.5μM的左西替利嗪和5μM的西替利津,以及5μM左西替利嗪和10μM西替利津之间的抑制作用是可比较的。此外,5μM左西替利嗪在抑制IL-8分泌方面优于5μM西替利津,但在其他条件下没有出现这种差异。我们的结果表明,更高浓度的西替利嗪和左西替利津可以减少IL-1β刺激的A549细胞释放GM-CSF和IL-8。这些观察结果表明,两种第二代抗组胺药的抗炎作用可能超过组胺H(1)受体拮抗剂,左西替利嗪在这种活性方面发挥着重要作用[2]。
|
| 细胞实验 |
细胞活力测定[2]
细胞类型:人气道上皮细胞系A549。 测试浓度:0-10 μM。 孵化持续时间:24小时。 实验结果:不同浓度的西替利嗪(0.1、1、2.5、5、10 μM)孵育24小时后,A549细胞的存活率均高于90%。通过MTT试验与对照组进行比较。 5 和 10 μM 西替利嗪分别抑制 GM-CSF 释放 70.71% 和 61.55%。与 10 μM 西替利嗪预孵育可抑制 IL -8 分泌 75.04%。 |
| 动物实验 |
Animal/Disease Models: Male 8weeks old BALB/ c mice (25-30 g) immunized and challenged with ragweed pollen[3].
Doses: 2 or 20 mg/kg. Route of Administration: Orally, diluted in sterile water on days 18, 19, and 20. Experimental Results: The neutrophilia at 8 h and eosinophilia at 24 h induced by ragweed pollen extract per os were Dramatically decreased in the mice treated with 20 mg/kg. The dosage with 2 mg/kg had no effect. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Cetirizine was rapidly absorbed with a time to maximum concentration (Tmax) of about 1 hour after oral administration of tablets or syrup formulation in adult volunteers. Bioavailability was found to be similar between the tablet and syrup dosage forms. When healthy study volunteers were given several doses of cetirizine (10 mg tablets once daily for 10 days), a mean peak plasma concentration (Cmax) of 311 ng/mL was measured. **Effect of food on absorption** Food had no effect on cetirizine exposure (AUC), however, Tmax was delayed by 1.7 hours and Cmax was decreased by 23% in the fed state. Mainly eliminated in the urine,. Between 70 – 85% of an orally administered dose can be found in the urine and 10 – 13% in the feces. Apparent volume of distribution: 0.44 +/- 0.19 L/kg. Apparent total body clearance: approximately 53 mL/min. Cetirizine is mainly eliminated by the kidneys,. Dose adjustment is required for patients with moderate to severe renal impairment and in patients on hemodialysis. A mass balance study in 6 healthy male volunteers indicated that 70% of the administered radioactivity was recovered in the urine and 10% in the feces. Approximately 50% of the radioactivity was identified in the urine as unchanged drug. Most of the rapid increase in peak plasma radioactivity was associated with parent drug, suggesting a low degree of first-pass metabolism. Cetirizine is metabolized to a limited extent by oxidative O-dealkylation to a metabolite with negligible antihistaminic activity. The enzyme or enzymes responsible for this metabolism have not been identified. The mean plasma protein binding of cetirizine is 93%, independent of concentration in the range of 25-1000 ng/mL, which includes the therapeutic plasma levels observed. Cetirizine was rapidly absorbed with a time to maximum concentration (Tmax) of approximately 1 hour following oral administration of tablets, chewable tablets or syrup in adults. Comparable bioavailability was found between the tablet and syrup dosage forms. Comparable bioavailability was also found between the Zyrtec tablet and the Zyrtec chewable tablet taken with or without water. When healthy volunteers were administered multiple doses of cetirizine (10 mg tablets once daily for 10 days), a mean peak plasma concentration (Cmax) of 311 ng/mL was observed. No accumulation was observed. Cetirizine pharmacokinetics were linear for oral doses ranging from 5 to 60 mg. Food had no effect on the extent of exposure (AUC) of the cetirizine tablet or chewable tablet, but Tmax was delayed by 1.7 hours and 2.8 hours respectively, and Cmax was decreased by 23% and 37%, respectively in the presence of food. When pediatric patients aged 7 to 12 years received a single, 5-mg oral cetirizine capsule, the mean Cmax was 275 ng/mL. Based on cross-study comparisons, the weight-normalized, apparent total body clearance was 33% greater and the elimination half-life was 33% shorter in this pediatric population than in adults. In pediatric patients aged 2 to 5 years who received 5 mg of cetirizine, the mean Cmax was 660 ng/mL. Based on cross-study comparisons, the weight-normalized apparent total body clearance was 81 to 111% greater and the elimination half-life was 33 to 41% shorter in this pediatric population than in adults. In pediatric patients aged 6 to 23 months who received a single dose of 0.25 mg/kg cetirizine oral solution (mean dose 2.3 mg), the mean Cmax was 390 ng/mL. Based on cross-study comparisons, the weight-normalized, apparent total body clearance was 304% greater and the elimination half-life was 63% shorter in this pediatric population compared to adults. The average AUC(0-t) in children 6 months to <2 years of age receiving the maximum dose of cetirizine solution (2.5 mg twice a day) is expected to be two-fold higher than that observed in adults receiving a dose of 10 mg cetirizine tablets once a day. For more Absorption, Distribution and Excretion (Complete) data for Cetirizine (7 total), please visit the HSDB record page. Metabolism / Metabolites A mass balance clinical trial comprised of 6 healthy male study volunteers showed that 70% of the administered radioactivity was measured in the urine and 10% in the feces after cetirizine administration. About 50% of the radioactivity was measured in the urine as unchanged cetirizine. Most of the rapid increase in peak plasma radioactivity was related to the parent drug, implying a low level of first pass metabolism. This prevents potential interactions of cetirizine with drugs interacting with hepatic cytochrome enzymes. Cetirizine is metabolized partially by oxidative O-dealkylation to a metabolite with insignificant antihistaminic activity. The enzyme or enzymes responsible for this step in cetirizine metabolism have not yet been identified. A mass balance study in 6 healthy male volunteers indicated that 70% of the administered radioactivity was recovered in the urine and 10% in the feces. Approximately 50% of the radioactivity was identified in the urine as unchanged drug. Most of the rapid increase in peak plasma radioactivity was associated with parent drug, suggesting a low degree of first-pass metabolism. Cetirizine is metabolized to a limited extent by oxidative O-dealkylation to a metabolite with negligible antihistaminic activity. The enzyme or enzymes responsible for this metabolism have not been identified. Pharmacokinetic parameters of hydroxyzine and its active metabolite cetirizine were determined after oral and intravenous administration of 2 mg kg(-1) of hydroxyzine to six healthy dogs. Plasma drug levels were determined with high-pressure liquid chromatography. Pharmacodynamic studies evaluated the suppressive effect on histamine and anticanine IgE-mediated cutaneous wheal formation. Pharmacokinetic and pharmacodynamic correlations were determined with computer modelling. The mean systemic availability of oral hydroxyzine was 72%. Hydroxyzine was rapidly converted to cetirizine regardless of the route of administration. The mean area-under-the-curve was eight and ten times higher for cetirizine than hydroxyzine after intravenous and oral dosing, respectively. After oral administration of hydroxyzine, the mean peak concentration of cetirizine was approximately 2.2 microg mL(-1) and that of hydroxyzine 0.16 ug mL(-1). The terminal half-life for cetirizine varied between 10 and 11 hr after intravenous and oral administration of hydroxyzine. A sigmoidal relationship was fit to the data comparing cetirizine plasma concentration to wheal suppression. Maximum inhibition (82% and 69% for histamine and anticanine IgE-mediated skin reactions, respectively) was observed during the first 8 hr, which correlated with a plasma concentration of cetirizine greater than 1.5 ug mL(-1). Pharmacological modelling suggested that increasing either hydroxyzine dosages or frequencies of administration would not result in histamine inhibition superior to that obtained with twice daily hydroxyzine at 2 mg kg(-1). In conclusion, there was rapid conversion of hydroxyzine to cetirizine. The reduction of wheal formation appeared almost entirely due to cetirizine. Pharmacodynamic modelling predicted that maximal antihistamine effect would occur with twice daily oral administration of hydroxyzine at 2 mg kg(-1). Half Life: 8.3 hours Biological Half-Life Plasma elimination half-life is 8.3 hours. The mean elimination half-life in 146 healthy volunteers across multiple pharmacokinetic studies was 8.3 hours ... . The pharmacokinetics of the second generation H1-receptor antagonist cetirizine were studied in 15 infants and toddlers (mean +/- SD age, 12.3 +/- 5.4 months) who were treated with a single 0.25 mg/kg dose of cetirizine solution. ... The elimination half-life was 3.1 +/- 1.8 hours ... |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
Cetirizine competes with histamine for binding at H1-receptor sites on the effector cell surface, resulting in suppression of histaminic edema, flare, and pruritus. The low incidence of sedation can be attributed to reduced penetration of cetirizine into the CNS as a result of the less lipophilic carboxyl group on the ethylamine side chain. Interactions A 72-year-old woman with renal insufficiency who was taking oral pilsicainide (150 mg/d) complained of feeling faint 3 days after she was prescribed oral cetirizine (20 mg/d). She was found to have a wide QRS wave with bradycardia. Her symptoms were relieved by termination of pilsicainide. The plasma concentrations of both drugs were significantly increased during the coadministration, and the cetirizine concentration decreased on cessation of pilsicainide despite the fact that treatment with cetirizine was continued, which suggested that the fainting was induced by the pharmacokinetic drug interaction. A pharmacokinetic study in 6 healthy male volunteers after a single dose of either cetirizine (20 mg) or pilsicainide (50 mg), or both, found that the renal clearance of each drug was significantly decreased by the coadministration of the drugs (from 475 +/- 101 mL/min to 279 +/- 117 mL/min for pilsicainide and from 189 +/- 37 mL/min to 118 +/- 28 mL/min for cetirizine; P = .008 and .009, respectively). In vitro studies using Xenopus oocytes with microinjected human organic cation transporter 2 and renal cells transfected with human multidrug resistance protein 1 revealed that the transport of the substrates of these transporters was inhibited by either cetirizine or pilsicainide. Thus elevated concentrations of these drugs as a result of a pharmacokinetic drug-drug interaction via either human multidrug resistance protein 1 or human organic cation transporter 2 (or both) in the renal tubular cells might have caused the arrhythmia in our patient. Although cetirizine has less potential for causing arrhythmias than other histamine 1 blockers, such an interaction should be considered, especially in patients with renal insufficiency who are receiving pilsicainide. The case of an 88-yr-old man who developed an increase in anticoagulant activity while taking cetirizine and acenocoumarol together is reported. The patient had been treated for 1 yr with acenocoumarol 1 mg daily for deep vein thrombosis. Three laboratory tests performed during the last 8 wk before hospitalization revealed prothrombin values of 58, 59, and 59% with an international normalized ratio (INR) of 1.5. After an accidental fall, the patient presented to the emergency department with epistaxis, which was severe and did not stop with the usual therapy. Laboratory tests revealed serum total protein 7.5 g/dL, serum creatinine 1.4 mg/dL, and platelet count 564x103/cu mm; the prothrombin was less than 10% with an INR of more than 14. Three days before admission, oral cetirizine 10 mg daily was prescribed for allergic rhinitis. On admission, cetirizine was stopped and the patient received factor IX complex. After the treatment the prothrombin increased to 100%. The patient was discharged 1 wk later with an INR of 1.42. The pharmacokinetics of the histamine H(1)-antagonist cetirizine and the effects of pretreatment with the antiparasitic macrocyclic lactone ivermectin on the pharmacokinetics of cetirizine were studied in horses. After oral administration of cetirizine at 0.2 mg/kg bw, the mean terminal half-life was 3.4 hr (range 2.9-3.7 hr) and the maximal plasma concentration 132 ng/mL (101-196 ng/mL). The time to reach maximal plasma concentration was 0.7 hr (0.5-0.8 hr). Ivermectin (0.2 mg/kg bw) given orally 1.5 hr before cetirizine did not affect its pharmacokinetics. However, ivermectin pretreatment 12 hr before cetirizine increased the area under the plasma concentration-time curve by 60%. The maximal plasma concentration, terminal half-life and mean residence time also increased significantly following the 12 hr pretreatment. Ivermectin is an inhibitor of P-glycoprotein, which is a major drug efflux transporter in cellular membranes at various sites. The elevated plasma levels of cetirizine following the pretreatment with ivermectin may mainly be due to decreased renal secretion, related to inhibition of the P-glycoprotein in the proximal tubular cells of the kidney. ... |
| 参考文献 | |
| 其他信息 |
Therapeutic Uses
Cetirizine alone or in fixed combination with pseudoephedrine hydrochloride is used for self-medication to provide symptomatic relief of seasonal allergic rhinitis (e.g., hay fever) or other upper respiratory allergies. Cetirizine alone or in fixed combination with pseudoephedrine hydrochloride also is used for the symptomatic treatment of perennial allergic rhinitis. It is recommended that the fixed combination generally be used only when both the antihistamine and nasal decongestant activity of the combination preparation are needed concurrently. /Included in US product label/ Cetirizine is used for self-medication to provide symptomatic relief of pruritus associated with chronic idiopathic urticaria (e.g., hives). /Included in US product label/ MEDICATION (VET): The efficacy of antihistamines for the treatment of pruritus is highly variable. The most commonly used antihistamines include ... cetirizine hydrochloride ... . /Cetirizine hydrochloride/ Drug Warnings In a controlled study of 1 week's duration in patients 6-11 months of age, those receiving cetirizine exhibited greater irritability/fussiness than those receiving placebo. In a controlled study in patients 12 months of age and older, insomnia occurred more frequently with cetirizine than with placebo (9 vs 5.3%, respectively). In those who received 5 mg or more daily, fatigue occurred in 3.6 or 1.3% and malaise in 3.5 or 1.8% of those receiving cetirizine or placebo, respectively. Fatigue or dizziness occurred in 5.9 or 2%, respectively, of patients 12 years of age and older receiving cetirizine, whereas these effects occurred in 2.6 or 1.2%, respectively, of patients receiving placebo. Headache was reported in more than 2% of patients 12 years of age and older receiving the drug; however, headache occurred more frequently in patients receiving placebo. In clinical trials in patients 6-11 years of age, headache occurred in 11, 14, or 12.3% of patients receiving 5-mg doses, 10-mg doses, or placebo, respectively. Abnormal coordination, ataxia, confusion, abnormal thinking, agitation, amnesia, anxiety, depersonalization, depression, emotional lability, euphoria, impaired concentration, insomnia, sleep disorders, nervousness, paroniria, dysphonia, asthenia, malaise, pain, hyperesthesia, hypoesthesia, hyperkinesia, hypertonia, migraine headache, myelitis, paralysis, paresthesia, ptosis, syncope, tremor, twitching, and vertigo have been reported in less than 2% of patients 12 years of age and older and children 6-11 years of age receiving cetirizine hydrochloride; however, a causal relationship to the drug has not been established. Aggressive reaction, seizures, hallucinations, suicidal ideation, and suicide have been reported rarely during postmarketing surveillance. The most frequent adverse effect in patients 12 years of age and older reported during cetirizine therapy is somnolence, occurring in 11, 14, or 6% of patients receiving 5-mg doses, 10-mg doses, or placebo, respectively. Overall, somnolence has been reported in 13.7 or 6.3% of patients receiving cetirizine or placebo, respectively. In addition, in clinical trials in patients 6-11 years of age, somnolence occurred in 1.9, 4.2, or 1.3% of patients receiving 5-mg doses, 10-mg doses, or placebo, respectively. Discontinuance of therapy because of somnolence has been reported in 1 or 0.6% of patients receiving cetirizine or placebo, respectively.1 3 In patients 6-24 months of age, somnolence occurred with essentially the same frequency in those who received cetirizine versus placebo. Adverse effects reported in 1% or more of patients 12 years of age and older with seasonal allergic rhinitis who received extended-release tablets of cetirizine hydrochloride in fixed combination with pseudoephedrine hydrochloride (Zyrtec-D) included insomnia, dry mouth, fatigue, somnolence, pharyngitis, epistaxis, accidental injury, dizziness, and sinusitis. For more Drug Warnings (Complete) data for Cetirizine (31 total), please visit the HSDB record page. Pharmacodynamics **General effects and respiratory effects** Cetirizine, the active metabolite of the piperazine H1-receptor antagonist hydroxyzine, minimizes or eliminates the symptoms of chronic idiopathic urticaria, perennial allergic rhinitis, seasonal allergic rhinitis, allergic asthma, physical urticaria, and atopic dermatitis. The clinical efficacy of cetirizine for allergic respiratory diseases has been well established in numerous trials. **Effects on urticaria/anti-inflammatory effects** It has anti-inflammatory properties that may play a role in asthma management. There is evidence that cetirizine improves symptoms of urticaria. Marked clinical inhibition of a wheal and flare response occurs in infants, children as well as adults within 20 minutes of one oral dose and lasts for 24 h. Concomitant use of cetirizine reduces the duration and dose of topical anti-inflammatory formulas used for the treatment of atopic dermatitis. |
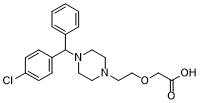
| 分子式 |
C21H25CLN2O3
|
|---|---|
| 分子量 |
388.888
|
| 精确质量 |
388.155
|
| 元素分析 |
C, 64.86; H, 6.48; Cl, 9.12; N, 7.20; O, 12.34
|
| CAS号 |
83881-51-0
|
| 相关CAS号 |
Cetirizine dihydrochloride;83881-52-1;Cetirizine-d4;1219803-84-5;Cetirizine-d8;774596-22-4;Levocetirizine;130018-77-8;Levocetirizine dihydrochloride;130018-87-0;Cetirizine methyl ester;83881-46-3;Cetirizine-d4 dihydrochloride;Cetirizine-d8 dihydrochloride;2070015-04-0
|
| PubChem CID |
2678
|
| 外观&性状 |
Crystals from ethanol
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
542.1±45.0 °C at 760 mmHg
|
| 熔点 |
110-115°C
|
| 闪点 |
281.6±28.7 °C
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
| 折射率 |
1.589
|
| LogP |
2.16
|
| tPSA |
53.01
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
443
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O=C(COCCN1CCN(C(C2C=CC(Cl)=CC=2)C2C=CC=CC=2)CC1)O
|
| InChi Key |
ZKLPARSLTMPFCP-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H25ClN2O3/c22-19-8-6-18(7-9-19)21(17-4-2-1-3-5-17)24-12-10-23(11-13-24)14-15-27-16-20(25)26/h1-9,21H,10-16H2,(H,25,26)
|
| 化学名 |
Acetic acid, (2-(4-((4-chlorophenyl)phenylmethyl)-1-piperazinyl)ethoxy)-
|
| 别名 |
Cetirizina; Cetryn; AC170; AC 170 AC-170; Ziptek; Setir; Virlix; Cetirizinum
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~250 mg/mL (~642.86 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5714 mL | 12.8571 mL | 25.7142 mL | |
| 5 mM | 0.5143 mL | 2.5714 mL | 5.1428 mL | |
| 10 mM | 0.2571 mL | 1.2857 mL | 2.5714 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































