| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
Chk1 (IC50 = 1.3 nM); Chk2 (IC50 = 2440 nM); ERK8 (IC50 = 130 nM); PKD1 (IC50 = 298 nM); RSK2 (IC50 = 361 nM); RSK1 (IC50 = 362 nM); FLT3 (IC50 = 582 nM); MARK3 (IC50 = 698 nM); NUAK1 (IC50 = 711 nM); CLK2 (IC50 = 2440 nM); BRSK1 (IC50 = 1660 nM); AMPK (IC50 = 2970 nM); PHK (IC50 = 3470 nM); CDK2/CyclA (IC50 = 3850 nM); CDK1/CyclB (IC50 = 9030 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:CCT245737 是重组人 CHK1 的有效抑制剂,IC50 为 1.4±0.3 nM(平均值±SD,n = 3,EZ Reader II 测定)。与功能重要的激酶 CDK1 和 CHK2 相比,CHK1 的选择性 > 1,000 倍(IC50 分别为 1.26-2.44 和 9.03 μM),对交叉反应激酶(如 ERK8、PKD1、RSK1)至少有 90 倍的选择性2. CCT245737 在多种人类肿瘤细胞系和人类肿瘤异种移植模型中有效抑制细胞 CHK1 活性 (IC50 30-220nM) 并增强吉西他滨和 SN38 细胞毒性。它可以消除依托泊苷诱导的 G2/M 期停滞。通过跨 CaCo2 细胞单层的转运测量,CCT245737 具有高细胞渗透性。激酶测定:CCT245737 显示 <50% 10= 121= 抑制= at= m= for= 代表= a= 选择性= chk1= of=> 5000 倍于这些酶。使用 10 μM CCT245737 在与激酶 Km、ATP 相对应的 ATP 浓度下针对 124 种人类激酶进行商业体外 33P 放射性激酶测定。 CHK2 和 FLT3 的其他激酶 IC50 测定是使用商业测定进行的,或在 LabChip® EZ Reader II 上使用重组人 CHK1 进行的,或在 DELFIA 测定中使用 CDK1 进行的。细胞测定:细胞毒性被确定为使用96小时(即4倍增)磺罗丹明B(SRB)测定对肿瘤细胞增殖(GI50)产生50%抑制的药物浓度。使用基于细胞的 ELISA 测量细胞内 CHK1 活性的抑制,以消除依托泊苷诱导的 G2 检查点(有丝分裂诱导测定,MIA)。
|
| 体内研究 (In Vivo) |
小鼠口服生物利用度是完全的(100%),且具有广泛的肿瘤暴露。 CCT245737 对 MYC 驱动的 B 细胞淋巴瘤小鼠模型显示出显着的单药活性。 BALB/c小鼠静脉注射10mg/kg CCT245737,血浆峰浓度为4μmol/L,半衰期为2.86h,AUC0-∞为9.96μmol.h/L,血浆清除率为2.1L /h/kg,分布容积大(0.19L)。等效口服剂量的曲线几乎相同,AUC0-∞为10.4μmol.h/L,显示出完全的口服生物利用度(F = 105%)。总之,CCT245737 显示出完全的口服生物利用度,具有线性药代动力学和与广泛的肿瘤暴露一致的高肿瘤/血浆比率。足够的 CCT245737 肿瘤药物暴露具有显着的抗肿瘤活性。
|
| 酶活实验 |
体外激酶测定[2]
使用10μM CCT245737,在与激酶Km、ATP相对应的ATP浓度下,对124种人激酶进行了商业化的体外33P放射性激酶测定。CHK2和FLT3的其他激酶IC50测定是使用商业试验(Z'-Lyte)或在LabChip®EZ Reader II(PerkinElmer)上使用重组人CHK1或在DELFIA试验中使用CDK1进行的 生化检测[1] 如前所述进行CHK1和CHK2抑制的体外试验。 CCT245737 (150 mg/kg po) 和 LY 188011 (100 mg/kg iv) 共同抑制 HT29 结肠癌异种移植物中的肿瘤生长。在 SW620 人结肠癌异种移植物中,CCT245737(300 mg/kg,口服)还可防止 LY 188011(60 mg/kg 静脉注射)24 小时后诱导的 pSer296 CHK1 自身磷酸化[1]。在人 B 细胞淋巴细胞白血病的 Eμ-Myc 小鼠模型中,CCT245737(150 mg/kg,口服)本身可显着抑制肿瘤生长[2]。 |
| 细胞实验 |
通过使用 96 小时磺罗丹明 B (SRB) 测定法来量化细胞毒性,以找到可抑制肿瘤细胞增殖 50% 的药物浓度 (GI50)。使用基于细胞的 ELISA,评估细胞内 CHK1 活性的抑制,以破坏 VP-16 诱导的 G2 检查点(有丝分裂诱导测定,MIA)。在存在诺考达唑的情况下,使用 UCN01 作为阳性对照来确定 G2 检查点废除 (MIA) IC50。化合物诱导有丝分裂的能力与其毒性相关,通过活性指数 (AI) 来衡量,即 MIA IC50 与 96 小时 SRB GI50 的比率。 CCT245737 的组合 GI50 通过使用固定浓度 (GI50) 的 SN38 或 LY 188011 与一系列 CCT245737 浓度组合进行标准增强研究来确定。增效指数(PI):CCT245737的单一GI50与CCT245737的组合GI50的比值表明CCT245737增强LY 188011或SN38细胞杀伤力的程度。 PI 值大于 1 表示遗传毒性活性增强。此外,为了确定与单独的基因毒性剂(即常规 PI)相比,CCT245737 增强药物细胞毒性的程度(单独的 GI50 基因毒性剂:GI50 基因毒性剂与无毒 CCT245737 浓度组合的比例,Con PI),使用以下方法进行了一系列实验:固定、无毒或最低毒性浓度的 CCT245737 (≤GI20) 与各种不同浓度的 LY 188011 或 SN38[2]。
|
| 动物实验 |
The female NCr athymic mice, aged 6-8 weeks, have s.c. injections of human HT29 colorectal carcinoma cells in their flanks. Dosing started five days after transplantation, or when the tumors reached a mean diameter of 5.5 mm. Compounds 4 (CCT245737) and 41 (150 mg/kg p.o.) are dosed in 10% DMSO 20% PEG 400, 5% Tween 80, and 65% water in the hours following each dose of LY 188011 (100 mg/kg i.v.). Days 0 through 14 are dosed in saline. Three times a week, body weights and tumor measurements are made. Tumors that grow to a predefined humane endpoint (mean diameter <15 mm) are individually culled from the animals[1].
Compound tolerability and pharmacokinetic investigations were carried out in female BALB/c mice (Charles River). Human tumor xenografts were established s.c. in female CRTac:Ncr-Fox1(nu) athymic mice and treated as previously described. The vehicle for oral administration of CCT245737 was DPTW (10% DMSO, 20% PEG400, 5% Tween 80 and 65% water) and gemcitabine and irinotecan were administered in their respective clinical vehicles. Treatments were generally initiated when tumors reached a mean diameter of 5-6mm (day 0). For combination studies, CCT245737 was given orally 24 and 48h after genotoxic drug administration, previously determined as an optimal schedule for CHK1 inhibitor and genotoxic drug combinations. In HT29 xenograft studies, gemcitabine was administered at 100mg/kg i.v. on days 0,7 and 14 and CCT245737 at the indicated doses on days 1,2,8,9,15 and16. Irinotecan was administered at 25mg/kg i.p. on days 0,4 and 8 with CCT245737 administered at 150mg/kg p.o. on days 1,2,5,6,9 and 10. In SW620 and Calu6 xenograft studies, gemcitabine was administered at 100mg/kg i.v. on days 0,4 and 8 and CCT245737 subsequently at 150mg/kg on days 1,2,5,6,9 and 10. For the Calu6 xenograft studies involving gemcitabine and carboplatin, drugs were administered at 100mg/kg i.v. and 5mg/kg i.p., respectively on day 0 with gemcitabine alone at 100mg/kg i.v. on day 7 with CCT245737 at 150mg/kg p.o. on days 1,2,8 and 9. The genotoxic drug doses employed were sub-maximally active to facilitate detection of subsequent potentiation. Initial treatment groups contained from 6 to 10 mice and animals were inspected daily and tumor size and volume measured every 2 or 3 days. Tumor volume and growth delay were determined as previously described [2]. Transgenic Eμ-Myc mice which develop aggressive infiltrating lymphoma were established and monitored as previously described. To generate transgenic Eμ-Myc driven lymphoma allografts, tumor cells from 3 separate tumors were harvested, cells counted and injected via a tail vein. Six mice were set up per tumor to provide 3 control and 3 treated animals, giving a maximum of 9 mice per treatment group. Animals were monitored daily and continuously using RFID transponders to measure temperature, activity and water consumption as previously described. For studies of single-agent CCT245737 activity in mice injected with transgenic Eμ-Myc lymphoid tumor cells, CCT245737 was administered at 150mg/kg p.o. for 9 successive days with culling 24h after the last dose. Lymph nodes and other tissues were removed from vehicle and CCT245737 treated mice and their weights and tissue/body weight ratios compared to assess antitumor activity. Bone marrow cellularity was also determined to check for tumor cell involvement. All mice were handled in compliance with local and national animal welfare guidelines[2]. |
| 参考文献 |
|
| 其他信息 |
Chk1 Inhibitor SRA737 is an orally bioavailable inhibitor of checkpoint kinase 1 (chk1), with potential antineoplastic and chemosensitization activities. Upon oral administration, chk1 inhibitor SRA737 selectively binds to chk1, thereby preventing chk1 activity and abrogating the repair of damaged DNA. This may lead to an accumulation of damaged DNA, inhibition of cell cycle arrest, and induction of apoptosis. SRA737 may potentiate the cytotoxicity of DNA-damaging agents and reverse tumor cell resistance to chemotherapeutic agents. Chk1, an adenosine triphosphate (ATP)-dependent serine/threonine kinase overexpressed in a variety of cancer cell types, mediates cell cycle checkpoint control and is essential for DNA repair; it plays a key role in resistance to chemotherapeutic agents by repairing DNA damage.
|
| 分子式 |
C16H16F3N7O
|
|
|---|---|---|
| 分子量 |
379.34
|
|
| 精确质量 |
379.136
|
|
| 元素分析 |
C, 50.66; H, 4.25; F, 15.02; N, 25.85; O, 4.22
|
|
| CAS号 |
1489389-18-5
|
|
| 相关CAS号 |
1489389-18-5;
|
|
| PubChem CID |
72165232
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 沸点 |
547.2±50.0 °C at 760 mmHg
|
|
| 闪点 |
284.7±30.1 °C
|
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
|
| 折射率 |
1.589
|
|
| LogP |
3.82
|
|
| tPSA |
108
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
11
|
|
| 可旋转键数目(RBC) |
5
|
|
| 重原子数目 |
27
|
|
| 分子复杂度/Complexity |
526
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
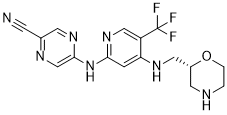
FC(F)(F)C(C=NC(NC1=CN=C(C#N)C=N1)=C2)=C2NC[C@H]3CNCCO3
|
|
| InChi Key |
YBYYWUUUGCNAHQ-LLVKDONJSA-N
|
|
| InChi Code |
InChI=1S/C16H16F3N7O/c17-16(18,19)12-8-25-14(26-15-9-22-10(4-20)5-24-15)3-13(12)23-7-11-6-21-1-2-27-11/h3,5,8-9,11,21H,1-2,6-7H2,(H2,23,24,25,26)/t11-/m1/s1
|
|
| 化学名 |
5-[[4-[[(2R)-morpholin-2-yl]methylamino]-5-(trifluoromethyl)pyridin-2-yl]amino]pyrazine-2-carbonitrile
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.59 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.59 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6362 mL | 13.1808 mL | 26.3616 mL | |
| 5 mM | 0.5272 mL | 2.6362 mL | 5.2723 mL | |
| 10 mM | 0.2636 mL | 1.3181 mL | 2.6362 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
|
|
|---|
|
 CHK1-IN-4 hydrochloride
CHK1-IN-4 hydrochloride
 PV1162
PV1162
 CHK1-IN-9
CHK1-IN-9
 Ziptide
Ziptide
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA





