| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
HDAC6 ( IC50 = 0.002 nM ); HDAC3 ( IC50 = 0.42 nM ); HDAC10 ( IC50 = 90.7 nM ); HDAC2 ( IC50 = 252 nM ); HDAC1 ( IC50 = 271 nM ); HDAC8 ( IC50 = 6851 nM )
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:CAY10603通过抑制HDAC6,对胰腺癌细胞系显示出有效的抗增殖活性,IC50<1 μM,可作为探索HDAC生物学的新分子探针激酶测定:纯化的HDAC与1 mM一起孵育将羧基荧光素 (FAM) 标记的乙酰化肽底物和测试化合物在含有 100 mM HEPES (pH 7.5)、25 mM KCl、0.1% BSA 和 0.01% Triton X-100 的 HDAC 测定缓冲液中于 25 °C 反应 17 小时。通过添加含有 0.078% SDS 的缓冲液来终止反应,最终 SDS 浓度为 0.05%。使用具有蓝色激光激发和绿色荧光检测 (CCD2) 的 Caliper LabChip 3000 系统通过电泳分离底物和产物。使用 Caliper 系统上的 Well Analyzer 软件测定底物和产物峰中的荧光强度。每个样品的反应重复进行。使用适用于 Microsoft Excel 的 IDBS XLFit 4.2.1 版插件和 XLFit 4 参数 Logistic 模型自动计算 IC50 值:((A+((B_A)/1+((C/x)D)))),其中x是化合物浓度,A和B分别是抑制百分比的估计最小值和最大值,C是拐点,D是S形曲线的希尔斜率。使用 Microsoft Excel 的 IDBS XLFit 版本 4.2.1 插件和公式 xf4_FitResultStdError 自动计算 IC50 值的标准误差。细胞测定:胰腺癌细胞系 BxPc-3、HupT3、Mia Paca-2、Panc 04.03 和 SU 86.86 在含有 10% 胎牛血清和 L-谷氨酰胺的培养基(DMEM 或 RPMI)中生长。将胰腺癌细胞一式两份放入 96 孔微量滴定板的 6 个孔中。电镀后四小时,用稀释剂 (DMSO) 或不同浓度的 SAHA 或指定的 HDACIs(浓度为 1 nM 至 50 mM)处理各个孔。根据制造商的建议,使用比色 MTT 测定在时间“0”和处理后 72 小时测量细胞毒性。 IC50 值使用 XLfit 计算。
|
| 体内研究 (In Vivo) |
HDAC6在AKI和CKD患者以及小鼠的肾小管上皮细胞(RTECs)中均显著上调。在LPS和腺嘌呤肾病诱导的AKI小鼠模型中,CAY10603具有显著的保护作用,包括改善生化指标和病理改变。体内和体外研究表明,CAY10603可有效抑制内质网应激源thapsigargin (Tg)触发的UPR中活化转录因子6 (ATF6)分支的激活。与这些发现一致,CAY10603在lps诱导的AKI和腺嘌呤诱导的肾病小鼠模型的RTECs中也显示出显著的ATF6激活抑制。结论:总的来说,这些结果表明CAY10603有望成为急性和慢性肾损伤的潜在治疗剂。[3]
|
| 酶活实验 |
将用 1 mm 羧基荧光素 (FAM) 标记的乙酰化肽底物和测试化合物与纯化的 HDAC 在含有 100 mm HEPES (pH 7.5)、25 mm KCl、0.1% 的 HDAC 测定缓冲液中于 25°C 孵育 17 小时BSA 和 0.01% Triton X-100。添加含有 0.078% SDS 的缓冲液,最终 SDS 浓度为 0.05%,反应结束。使用配备蓝色激光激发和绿色荧光检测 (CCD2) 的 Caliper LabChip 3000 系统,通过电泳分离底物和产物。 Caliper 系统的 Well Analyzer 软件用于计算底物和产物峰中的荧光强度。对于每个样品,反应均重复进行。 XLFit 4 参数 Logistic 模型(S 型剂量反应模型)和 Microsoft Excel 的 IDBS XLFit 版本 4.2.1 插件用于自动计算 IC50 值:((A+ ((B_A)/1+((C/ x)D)))),其中 x 是化合物浓度,A 和 B 分别表示估计的最小和最大抑制百分比,C 是 S 形曲线的拐点,D 是其山坡。
|
| 细胞实验 |
ATCC 提供胰腺癌细胞系 BxPc-3、HupT3、Mia Paca-2、Panc 04.03 和 SU 86.86。细胞系在补充有10%胎牛血清和L-谷氨酰胺的DMEM或RPMI培养基中培养。在 96 孔微量滴定板的 6 个孔中,以每孔 2.5-4P103 个细胞的密度铺板重复的胰腺癌细胞。电镀后四小时,用稀释剂 (DMSO)、不同浓度的 SAHA 或浓度为 1 nm 至 50 mm 的指定 HDACIs 处理各个孔。比色MTT测定用于测量处理后时间“0”和72小时的细胞毒性。 XLfit 用于计算 IC50 值。
|
| 动物实验 |
Histone deacetylase 6 (HDAC6) inhibitor CAY10603 has been identified as a potential therapeutic agent for the treatment of diabetic kidney disease (DKD). The objective of this study was to investigate the therapeutic effects of CAY10603 in mice with acute kidney injury (AKI) and chronic kidney diseases (CKD).
Methods: Renal immunohistology was performed to assess the expression levels of HDAC6 in both human and mouse kidney samples. C57BL/6J mice were intraperitoneal injected with lipopolysaccharide (LPS) to induce AKI; CD-1 mice were fed with adenine diet to induce adenine-nephropathy as CKD model. Serum creatinine, blood urea nitrogen and uric acid were measured to reflect renal function; renal histology was applied to assess kidney damage. Western blot and immunohistology were used to analyze the unfolded protein response (UPR) level.[3]
|
| 参考文献 |
|
| 其他信息 |
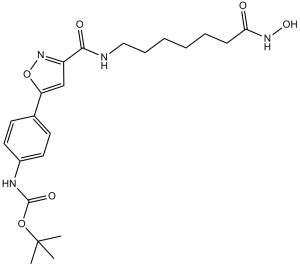
N-[4-[3-[[[7-(hydroxyamino)-7-oxoheptyl]amino]-oxomethyl]-5-isoxazolyl]phenyl]carbamic acid tert-butyl ester is a carbamate ester.
A series of hydroxamate based HDAC inhibitors containing a phenylisoxazole as the CAP group has been synthesized using nitrile oxide cycloaddition chemistry. An HDAC6 selective inhibitor having a potency of approximately 2 picomolar was identified. Some of the compounds were examined for their ability to block pancreatic cancer cell growth and found to be about 10-fold more potent than SAHA. This research provides valuable, new molecular probes for use in exploring HDAC biology.[1] Histone deacetylases (HDACs) are promising targets for cancer therapy, and first-generation HDAC inhibitors are currently in clinical trials for the treatment of cancer patients. HDAC6, which is a key regulator of many signaling pathways that are linked to cancer, has recently emerged as an attractive target for the treatment of cancer. In the present study, HDAC6 was found to be overexpressed in lung adenocarcinoma cell lines and was negatively correlated with the prognosis of patients with lung adenocarcinoma. Overexpression of HDAC6 promoted the proliferation of lung adenocarcinoma cells in a deacetylase activity-dependent manner. HDAC6 overexpression conferred resistance to gefitinib via the stabilization of epidermal growth factor receptor (EGFR). The inhibition of HDAC6 by CAY10603, a potent and selective inhibitor of HDAC6, inhibited the proliferation of lung adenocarcinoma cells and induced apoptosis. CAY10603 downregulated the levels of EGFR protein, which in turn inhibited activation of the EGFR signaling pathway. Moreover, CAY10603 synergized with gefitinib to induce apoptosis of the lung adenocarcinoma cell lines via the destabilization of EGFR. Taken together, our results suggest that the inhibition of HDAC6 may be a promising strategy for the treatment of lung adenocarcinoma.[2] A series of 2-phenylthiazole analogues were designed and synthesized as potential histone deacetylase 6 (HDAC6) inhibitors based on compound 12c (an HDAC6/tubulin dual inhibitor discovered by us recently) and CAY10603 (a known HDAC6 inhibitor). Among them, compound XP5 was the most potent HDAC6 inhibitor with an IC50 of 31 nM and excellent HDAC6 selectivity (SI = 338 for HDAC6 over HDAC3). XP5 also displayed high antiproliferative activity against various cancer cell lines including the HDACi-resistant YCC3/7 gastric cancer cells (IC50 = 0.16-2.31 μM), better than CAY10603. Further, XP5 (50 mg/kg) exhibited significant antitumor efficacy in a melanoma tumor model with a tumor growth inhibition (TGI) of 63% without apparent toxicity. Moreover, XP5 efficiently enhanced the in vivo antitumor immune response when combined with a small-molecule PD-L1 inhibitor, as demonstrated by the increased tumor-infiltrating lymphocytes and reduced PD-L1 expression levels. Taken together, the above results suggest that XP5 is a promising HDAC6 inhibitor deserving further investigation.J Med Chem. 2022 Feb 10;65(3):2434-2457. |
| 分子式 |
C22H30N4O6
|
|
|---|---|---|
| 分子量 |
446.5
|
|
| 精确质量 |
446.216
|
|
| 元素分析 |
C, 59.18; H, 6.77; N, 12.55; O, 21.50
|
|
| CAS号 |
1045792-66-2
|
|
| 相关CAS号 |
|
|
| PubChem CID |
24951314
|
|
| 外观&性状 |
White to light yellow solid powder
|
|
| 密度 |
1.2±0.1 g/cm3
|
|
| 折射率 |
1.563
|
|
| LogP |
1.94
|
|
| tPSA |
142.79
|
|
| 氢键供体(HBD)数目 |
4
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
12
|
|
| 重原子数目 |
32
|
|
| 分子复杂度/Complexity |
616
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O(C(N([H])C1C([H])=C([H])C(=C([H])C=1[H])C1=C([H])C(C(N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(N([H])O[H])=O)=O)=NO1)=O)C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H]
|
|
| InChi Key |
WWGBHDIHIVGYLZ-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C22H30N4O6/c1-22(2,3)31-21(29)24-16-11-9-15(10-12-16)18-14-17(26-32-18)20(28)23-13-7-5-4-6-8-19(27)25-30/h9-12,14,30H,4-8,13H2,1-3H3,(H,23,28)(H,24,29)(H,25,27)
|
|
| 化学名 |
tert-butyl N-[4-[3-[[7-(hydroxyamino)-7-oxoheptyl]carbamoyl]-1,2-oxazol-5-yl]phenyl]carbamate
|
|
| 别名 |
CAY-10603; CAY10603; BML-281; tert-Butyl (4-(3-((7-(hydroxyamino)-7-oxoheptyl)carbamoyl)isoxazol-5-yl)phenyl)carbamate; CAY-10603; tert-butyl N-[4-[3-[[7-(hydroxyamino)-7-oxoheptyl]carbamoyl]-1,2-oxazol-5-yl]phenyl]carbamate; CHEMBL511749; compound 3 [PMID: 18642892]; CAY 10603
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.60 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.60 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.60 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 5% DMSO+50% PEG 300+ddH2O: 9mg/mL 配方 5 中的溶解度: 5 mg/mL (11.20 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液; 需要超声波加热并加热至 42°C。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2396 mL | 11.1982 mL | 22.3964 mL | |
| 5 mM | 0.4479 mL | 2.2396 mL | 4.4793 mL | |
| 10 mM | 0.2240 mL | 1.1198 mL | 2.2396 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
|
 HDAC6-IN-18
HDAC6-IN-18
 Mz325
Mz325
 WW437
WW437
 PI3Kα/HDAC6-IN-1
PI3Kα/HDAC6-IN-1
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA



