| 规格 | 价格 | |
|---|---|---|
| 10mg | ||
| 500mg |

| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Systemic absorption of local anesthetics is dose- and concentration-dependendent on the total drug administered. Other factors that affect the rate of systemic absorption include the route of administration, blood flow at the administration site, and the presence or absence of epinephrine in the anesthetic solution. Bupivacaine formulated for instillation with [meloxicam] produced varied systemic measures following a single dose of varying strength. In patients undergoing bunionectomy, 60 mg of bupivacaine produced a Cmax of 54 ± 33 ng/mL, a median Tmax of 3 h, and an AUC∞ of 1718 ± 1211 ng\*h/mL. For a 300 mg dose used in herniorrhaphy, the corresponding values were 271 ± 147 ng/mL, 18 h, and 15,524 ± 8921 ng\*h/mL. Lastly, a 400 mg dose used in total knee arthroplasty produced values of 695 ± 411 ng/mL, 21 h, and 38,173 ± 29,400 ng\*h/mL. Only 6% of bupivacaine is excreted unchanged in the urine. After absorption into the blood, bupivacaine hydrochloride is more highly bound to plasma proteins than are any other local anesthetics; bupivacaine is reportedly 82-96% bound. Bupivacaine hydrochloride has the lowest degree of placental transmission of parenteral local anesthetics and may cause the least fetal depression. Pregnant rats received an intravenous infusion of bupivacaine at a rate of 0.33 mg. kg-1. min-1 over a period of 15 min. The fetuses were delivered either at the end of infusion or at 2 or 4 hr after dosing. Maternal and fetal blood and tissue samples were obtained for the assays of bupivacaine and its metabolites using capillary gas chromatography-mass spectrometry. The elimination half-life of bupivacaine was 37.7 min. The major metabolite was 3'-hydroxybupivacaine. Bupivacaine and 3'-hydroxybupivacaine were present in all samples at the end of administration. The fetal to maternal concentration ratio of bupivacaine in plasma was 0.29, and in the placenta was 0.63. The amnion contained the highest bupivacaine concentration: threefold higher in the maternal and 11-fold higher than in the fetal plasma. At 4 hr after dosing, bupivacaine was no longer detectable in any maternal and fetal samples, whereas 3'-hydroxybupivacaine was still present in all tissues except the fetal plasma and heart. These data indicate that a considerable amount of bupivacaine is taken up by both sides of the placenta, as well as the amnion and myometrium. 3'-Hydroxybupivacaine was present in all tissues except the fetal plasma and heart samples, even after the parent compound became no longer detectable. After injection of Bupivacaine Hydrochloride for caudal, epidural, or peripheral nerve block in man, peak levels of bupivacaine in the blood are reached in 30 to 45 minutes, followed by a decline to insignificant levels during the next three to six hours. Pharmacokinetic studies on the plasma profile of Bupivacaine Hydrochloride after direct intravenous injection suggest a three-compartment open model. The first compartment is represented by the rapid intravascular distribution of the drug. The second compartment represents the equilibration of the drug throughout the highly perfused organs such as the brain, myocardium, lungs, kidneys, and liver. The third compartment represents an equilibration of the drug with poorly perfused tissues, such as muscle and fat. The elimination of drug from tissue distribution depends largely upon the ability of binding sites in the circulation to carry it to the liver where it is metabolized. For more Absorption, Distribution and Excretion (Complete) data for Bupivacaine (6 total), please visit the HSDB record page. Metabolism / Metabolites Amide-type local anesthetics such as bupivacaine are metabolized primarily in the liver via conjugation with glucuronic acid. The major metabolite of bupivacaine is 2,6-pipecoloxylidine, which is mainly catalyzed via cytochrome P450 3A4. Pregnant rats received an intravenous infusion of bupivacaine at a rate of 0.33 mg. kg-1. min-1 over a period of 15 min. The fetuses were delivered either at the end of infusion or at 2 or 4 hr after dosing. Maternal and fetal blood and tissue samples were obtained for the assays of bupivacaine and its metabolites using capillary gas chromatography-mass spectrometry. The elimination half-life of bupivacaine was 37.7 min. The major metabolite was 3'-hydroxybupivacaine. Bupivacaine and 3'-hydroxybupivacaine were present in all samples at the end of administration. The fetal to maternal concentration ratio of bupivacaine in plasma was 0.29, and in the placenta was 0.63. The amnion contained the highest bupivacaine concentration: threefold higher in the maternal and 11-fold higher than in the fetal plasma. At 4 hr after dosing, bupivacaine was no longer detectable in any maternal and fetal samples, whereas 3'-hydroxybupivacaine was still present in all tissues except the fetal plasma and heart. These data indicate that a considerable amount of bupivacaine is taken up by both sides of the placenta, as well as the amnion and myometrium. 3'-Hydroxybupivacaine was present in all tissues except the fetal plasma and heart samples, even after the parent compound became no longer detectable. Bupivacaine hydrochloride is principally metabolized to pipecolylxylidine (PPX) by N-dealkylation, probably in the liver. Bupivacaine is excreted in urine as small amounts of PPX, unchanged drug (5%), and other metabolites as yet unidentified. Amide-type local anesthetics such as bupivacaine are metabolized primarily in the liver via conjugation with glucuronic acid. The major metabolite of bupivacaine is 2,6-pipecoloxylidine, which is mainly catalyzed via cytochrome P450 3A4. Route of Elimination: Only 6% of bupivacaine is excreted unchanged in the urine. Half Life: 2.7 hours in adults and 8.1 hours in neonates Biological Half-Life 2.7 hours in adults and 8.1 hours in neonates. Bupivacaine applied together with [meloxicam] for postsurgical analgesia had a median half-life of 15-17 hours, depending on dose and application site. Pregnant rats received an intravenous infusion of bupivacaine at a rate of 0.33 mg. kg-1. min-1 over a period of 15 min. The fetuses were delivered either at the end of infusion or at 2 or 4 hr after dosing. Maternal and fetal blood and tissue samples were obtained for the assays of bupivacaine and its metabolites using capillary gas chromatography-mass spectrometry. The elimination half-life of bupivacaine was 37.7 min. The elimination half-life of bupivacaine hydrochloride is 1.5-5.5 hours in adults and 8.1 hours in neonates. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation Because of the low levels of bupivacaine in breastmilk, and it is not orally absorbed, amounts received by the infant are small and it has not caused any adverse effects in breastfed infants. Bupivacaine during labor and delivery with other anesthetics and analgesics has been reported by some to interfere with breastfeeding. However, this assessment is controversial and complex because of the many different combinations of drugs, dosages and patient populations studied as well as the variety of techniques used and deficient design of many of the studies. In contrast, epidural bupivacaine begun after clamping of the umbilical cord appears to enhance breastfeeding success because of improved pain control. Overall, it appears that with good breastfeeding support epidural bupivacaine with or without fentanyl or one of its derivatives has little or no adverse effect on breastfeeding success. Labor pain medication may delay the onset of lactation. ◉ Effects in Breastfed Infants Bupivacaine administered to the mother by the epidural route for labor analgesia had no apparent adverse effect on 13 breastfed infants. Thirty patients who underwent cesarean section received a bilateral transverses abdominus plane block using of a mixture of 52 mg bupivacaine hydrochloride 0.25% and 266 mg liposomal bupivacaine 1.3%. Two of the infants had transient tachypnea, but causality could not be determined. None of the infants required hospital readmission during the 14-day follow-up period. ◉ Effects on Lactation and Breastmilk Thirty women who delivered by cesarean section received either spinal anesthesia (not defined) alone (n = 15) or spinal anesthesia plus bupivacaine (n = 15) by extradural infusion after clamping the umbilical cord. A bupivacaine bolus of 12.5 mg was followed by a continuous infusion of 17.5 mg/hour for 3 days postpartum. Patients who received bupivacaine had better pain relief as indicated by lower pain scores and a lower consumption of supplemental diclofenac for pain. Bupivacaine-treated patients also produced more milk per day than the untreated women, a difference that was statistically significant from day 3 to the end of the study on day 11 postpartum. The authors concluded that improved pain relief improved breastfeeding performance. Twenty women who delivered by cesarean section received either bupivacaine alone or bupivacaine plus buprenorphine by extradural infusion after clamping the umbilical cord. A bupivacaine bolus of 12.5 mg was followed by a continuous infusion of 17.5 mg/hour for 3 days. The buprenorphine was given as a bolus of 200 mcg followed by 8.4 mcg/hour for 3 days. Patients started breastfeeding as soon as they were able to sit up. Both the amount of milk fed and infant weight increased in both groups over the first 10 days postpartum; however, the increases were greater in those who received bupivacaine alone. A prospective cohort study compared women who received no analgesia (n = 63) to women who received continuous epidural analgesia with fentanyl and either bupivacaine 0.05 to 0.1% (n = 39) or ropivacaine (n = 13) during labor and delivery. The total dosage of bupivacaine was 31 to 62 mg and the average total infusion time from start to delivery was 219 minutes. The study found no differences between the groups in breastfeeding effectiveness or infant neurobehavioral status at 8 to 12 hours postpartum or the number exclusively or partially breastfeeding at 4 weeks postpartum. A randomized, prospective study measured infant breastfeeding behavior following epidural or intravenous fentanyl during delivery in 100 multiparous mothers undergoing cesarean section and delivering fullterm, healthy infants. The epidural group received epidural bupivacaine 100 mg initially, followed by a continuous infusion of 25 mg/hour. The intravenous fentanyl group received a spinal injection of 15 to 20 mg of bupivacaine. A slight difference was seen in breastfeeding behavior between the groups, with the infants in the intravenous fentanyl group performing slightly worse than those in the epidural group. However, all mothers were able to breastfeed their infants at 24 hours. None had severe breastfeeding problems; 10 women in the epidural group reported mild or moderate problems and 7 women in the intravenous group reported breastfeeding problems. Twenty mothers in the epidural group and 14 in the intravenous group used supplemental bottle feeding, with the difference not statistically significant. A randomized, but nonblinded, study in women undergoing cesarean section compared epidural anesthesia with bupivacaine to general anesthesia with intravenous thiopental 4 mg/kg and succinylcholine 1.5 mg/kg for induction followed by nitrous oxide and isoflurane. The time to the first breastfeed was significantly shorter (107 vs 228 minutes) with the epidural anesthesia than with general anesthesia. This difference was probably caused by the anesthesia's effects on the infant, because the Apgar and neurologic and adaptive scores were significantly lower in the general anesthesia group of infants. A randomized, multicenter trial compared the initiation rate and duration of breastfeeding in women who received high-dose epidural bupivacaine alone, or one of two low-dose combinations of bupivacaine plus fentanyl. A nonepidural matched control group was also compared. No differences in breastfeeding initiation rates or duration were found among the epidural and nonmedicated, nonepidural groups. A nonrandomized study in low-risk mother-infant pairs found that there was no difference overall in the amount of sucking by newborns, whether their mothers received bupivacaine plus fentanyl, or fentanyl alone by epidural infusion in various dosages, or received no analgesia for childbirth. In a subanalysis by sex and number of sucks, female infants were affected by high-dose bupivacaine and high-dose fentanyl, but male infant were not. However, the imbalance of many factors between the study groups makes this study difficult to interpret. In a prospective cohort study, 87 multiparous women who received epidural bupivacaine and fentanyl for pain control during labor and vaginal delivery. A loading dose of 0.125% bupivacaine with fentanyl 50-100 mcg. Epidural analgesia is maintained using 0.0625% bupivacaine and fentanyl 0.2 mcg/mL. The median dose of fentanyl received by the women was 151 mcg (range 30 to 570 mcg). The women completed questionnaires at 1 and 6 weeks postpartum regarding breastfeeding. Most women had prior experience with breastfeeding, support at home and ample time off from work. All women were breastfeeding at 1 week postpartum and 95.4% of women were breastfeeding at 6 weeks postpartum. A national survey of women and their infants from late pregnancy through 12 months postpartum compared the time of lactogenesis II in mothers who did and did not receive pain medication during labor. Categories of medication were spinal or epidural only, spinal or epidural plus another medication, and other pain medication only. Women who received medications from any of the categories had about twice the risk of having delayed lactogenesis II (>72 hours) compared to women who received no labor pain medication. A randomized study compared the effects of cesarean section using general anesthesia, spinal anesthesia, or epidural anesthesia, to normal vaginal delivery on serum prolactin and oxytocin as well as time to initiation of lactation. Spinal anesthesia used bupivacaine 10 to 11 mg of hypertonic 5% bupivacaine solution and epidural anesthesia used 10 mL (50 mg) of 0.5% bupivacaine. After delivery, patients in all groups received an infusion of oxytocin 30 international units in 1 L of saline, and 0.2 mg of methylergonovine if they were not hypertensive. Patients in the general anesthesia group (n = 21) had higher post-procedure prolactin levels and a longer mean time to lactation initiation (25 hours) than in the other groups (10.8 to 11.8 hours). Postpartum oxytocin levels in the nonmedicated vaginal delivery group were higher than in the general and spinal anesthesia groups and serum oxytocin in the epidural group were higher than those in the spinal group. A retrospective study in a Spanish public hospital compared the infants of mothers who received an epidural during labor that contained fentanyl and either bupivacaine or ropivacaine. Infants of mothers who received an epidural had a lower frequency of early breastfeeding. A randomized, double-blind study compared three epidural maintenance solutions for labor analgesia in women receiving epidural analgesia during labor: bupivacaine 1 mg/mL, bupivacaine 0.8 mg/mL with fentanyl 1 mcg/mL, or bupivacaine 0.625 mg/mL with fentanyl 2 mcg/mL. At 6 weeks postpartum, the breastfeeding rate was 94% or greater in all groups, with no difference among them. All mothers delivered full-term infants and were highly motivated to breastfeed and almost all had vaginal deliveries. A prospective cohort study in 1204 Israeli women on the effect of labor epidural analgesia during labor, the following protocol was used: bupivacaine 0.1% 15 mL and fentanyl 100 mcg in 5-mL increments, followed by an epidural infusion of bupivacaine 0.1% 10 mL and fentanyl 2 mcg/mL, with a patient-controlled epidural analgesia modality with 5 mL bolus with a lock-out time of 15 minutes. At 6 weeks postpartum, the breastfeeding and exclusive breastfeeding rates were lower (74% and 52%, respectively) in mothers who received the epidural analgesia than in those who did not (83% and 68%, respectively). However, the difference was mostly accounted for by parity, with the intervention having little effect on multiparous women. A retrospective study of women in a Turkish hospital who underwent elective cesarean section deliveries compared women who received bupivacaine spinal anesthesia (n = 170) to women who received general anesthesia (n = 78) with propofol for induction, sevoflurane for maintenance and fentanyl after delivery. No differences in breastfeeding rates were seen between the groups at 1 hour and 24 hours postpartum. However, at 6 months postpartum, 67% of women in the general anesthesia group were still breastfeeding compared to 81% in the spinal anesthesia group, which was a statistically significant difference. A study of 169 pregnant women randomized them to receive one of three solutions as epidural anesthesia during labor. Bupivacaine 0.1% or 0.125% was combined with sufentanil 5 mcg and bupivacaine 0.1% was combined with sufentanil 10 mcg, each in 15 mL. No difference in average LATCH score was found among the infants in the 3 groups. An observational study in Sweden compared nursing behaviors of the infants of mothers who received intravenous oxytocin or intramuscular oxytocin with or without receiving epidural analgesia with sufentanil (median dose 10 mcg) and bupivacaine (median dose 17.5 mg). Infants of mothers who received oxytocin infusions alone during labor breastfed as well as those of mothers who had no interventions during labor. Mothers who received oxytocin plus epidural analgesia had reduced breastfeeding behaviors and more weight loss at 2 days postpartum than those who did not receive epidural analgesia. The mothers of infants who breastfed well had greater variability in serum oxytocin than those whose infants did not breastfeed well. A nonrandomized, nonblinded study in a Serbian hospital of women near term who underwent cesarean section compared general anesthesia (n = 284) to spinal or epidural anesthesia (n = 249). Spinal anesthesia consisted of hyperbaric bupivacaine 12 mg and fentanyl 0.01 mg; epidural anesthesia consisted of isobaric bupivacaine 0.5% (0.5 mg per 10 cm height) and fentanyl 0.05 mg. General anesthesia consisted of propofol 2.3 mg/kg and succinylcholine 1.5 mg/kg for induction and intubation, followed by an anesthetic gas mixture and oxygen. Reportedly, nitric oxide (possibly nitrous oxide) was 50% of the gas before delivery and 67% after delivery. Sevoflurane was also used in some cases. After delivery and cord clamping, mothers received fentanyl 3 mcg/kg and rocuronium 0.5 mg/kg intravenously for placental delivery. After surgery, neuromuscular block reversal was performed with neostigmine and atropine. All patients received 1 mg/kg of diclofenac every 8 h for 24 hours after delivery and 98% of general anesthesia patients also received 100 mg of tramadol and 78.5% received acetaminophen 1 gram. No regional anesthesia patients received tramadol or acetaminophen. Patients receiving one of the regional anesthetic protocols established lactation sooner (56% and 29% after 18 and 24 hours, respectively), while 86% of women receiving general anesthesia did not establish lactation until 36 to 48 hours after surgery. Protein Binding Bupivacaine is ~95% protein bound. |
| 参考文献 |
|
| 其他信息 |
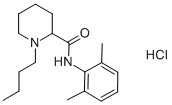
1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide is a piperidinecarboxamide obtained by formal condensation of the carboxy group of N-butylpipecolic acid with the amino group of 2,6-dimethylaniline. It is a piperidinecarboxamide, an aromatic amide and a tertiary amino compound. It is a conjugate base of a 1-butyl-2-[(2,6-dimethylphenyl)carbamoyl]piperidinium.
Bupivacaine is a widely used local anesthetic agent. Bupivacaine is an Amide Local Anesthetic. The physiologic effect of bupivacaine is by means of Local Anesthesia. Bupivacaine is an amide-type, long-acting local anesthetic. Bupivicaine reversibly binds to specific sodium ion channels in the neuronal membrane, resulting in a decrease in the voltage-dependent membrane permeability to sodium ions and membrane stabilization; inhibition of depolarization and nerve impulse conduction; and a reversible loss of sensation. Liposomal Bupivacaine is a liposome-encapsulated formulation of bupivacaine, which is an amide-type, long-acting local anesthetic. Upon administration, bupivacaine reversibly binds to specific sodium ion channels in the neuronal membrane, resulting in both a decrease in the voltage-dependent membrane permeability to sodium ions and membrane stabilization. This leads to inhibition of both depolarization and nerve impulse conduction, and a reversible loss of sensation. Compared to bupivacaine alone, liposomal delivery increases the duration of local anesthetic action and delays the peak plasma concentration of bupivacaine due to its slow release from the liposome. Bupivacaine is only found in individuals that have used or taken this drug. It is a widely used local anesthetic agent. Bupivacaine blocks the generation and the conduction of nerve impulses, presumably by increasing the threshold for electrical excitation in the nerve, by slowing the propagation of the nerve impulse, and by reducing the rate of rise of the action potential. Bupivacaine binds to the intracellular portion of sodium channels and blocks sodium influx into nerve cells, which prevents depolarization. In general, the progression of anesthesia is related to the diameter, myelination and conduction velocity of affected nerve fibers. Clinically, the order of loss of nerve function is as follows: (1) pain, (2) temperature, (3) touch, (4) proprioception, and (5) skeletal muscle tone. The analgesic effects of Bupivicaine are thought to potentially be due to its binding to the prostaglandin E2 receptors, subtype EP1 (PGE2EP1), which inhibits the production of prostaglandins, thereby reducing fever, inflammation, and hyperalgesia. A widely used local anesthetic agent. See also: Bupivacaine; meloxicam (component of). Drug Indication As an implant, bupivacaine is indicated in adults for placement into the surgical site to produce postsurgical analgesia for up to 24 hours following open inguinal hernia repair. Bupivacaine, in liposome suspension, is indicated in patients aged 6 years and older for single-dose infiltration to produce postsurgical local analgesia. In adults, it is also indicated as an interscalene brachial plexus nerve block to produce postsurgical regional analgesia. Bupivacaine, in combination with [meloxicam], is indicated for postsurgical analgesia in adult patients for up to 72 hours following foot and ankle, small-to-medium open abdominal, and lower extremity total joint arthroplasty surgical procedures. Bupivacaine, alone or in combination with [epinephrine], is indicated in adults for the production of local or regional anesthesia or analgesia for surgery, dental and oral surgery procedures, diagnostic and therapeutic procedures, and for obstetrical procedures. Specific concentrations and presentations are recommended for each type of block indicated to produce local or regional anesthesia or analgesia. Finally, its use is not indicated in all blocks given clinically significant risks associated with use. FDA Label Exparel liposomal is indicated: in adults as a brachial plexus block or femoral nerve block for treatment of post-operative pain. in adults and children aged 6 years or older as a field block for treatment of somatic post-operative pain from small- to medium-sized surgical wounds. Postsurgical analgesia Mechanism of Action Like [lidocaine], bupivacaine is an amide local anesthetic that provides local anesthesia through blockade of nerve impulse generation and conduction. These impulses, also known as action potentials, critically depend on membrane depolarization produced by the influx of sodium ions into the neuron through voltage-gated sodium channels. Bupivacaine crosses the neuronal membrane and exerts its anesthetic action through blockade of these channels at the intracellular portion of their pore-forming transmembrane segments. The block is use-dependent, where repetitive or prolonged depolarization increases sodium channel blockade. Without sodium ions passing through the channel’s pore, bupivacaine stabilizes the membrane at rest and therefore prevents neurotransmission. In general, the progression of anesthesia is related to the diameter, myelination and conduction velocity of affected nerve fibers. Clinically, the order of loss of nerve function is as follows: (1) pain, (2) temperature, (3) touch, (4) proprioception, and (5) skeletal muscle tone. While it is well-established that the main action of bupivacaine is through sodium channel block, additional analgesic effects of bupivacaine are thought to potentially be due to its binding to the prostaglandin E2 receptors, subtype EP1 (PGE2EP1), which inhibits the production of prostaglandins, thereby reducing fever, inflammation, and hyperalgesia. Local anesthetics block the generation and the conduction of nerve impulses, presumably by increasing the threshold for electrical excitation in the nerve, by slowing the propagation of the nerve impulse, and by reducing the rate of rise of the action potential. In general, the progression of anesthesia is related to the diameter, myelination, and conduction velocity of affected nerve fibers. Clinically, the order of loss of nerve function is as follows: (1) pain, (2) temperature, (3) touch, (4) proprioception, and (5) skeletal muscle tone. |
| 分子式 |
C18H29CLN2O
|
|---|---|
| 分子量 |
324.8887
|
| 精确质量 |
324.196
|
| CAS号 |
14252-80-3
|
| PubChem CID |
2474
|
| 外观&性状 |
White to off-white solid powder
|
| 沸点 |
423.4ºC at 760 mmHg
|
| 熔点 |
249-251ºC
|
| 闪点 |
209.9ºC
|
| 蒸汽压 |
2.24E-07mmHg at 25°C
|
| LogP |
4.709
|
| tPSA |
32.34
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
21
|
| 分子复杂度/Complexity |
321
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
LEBVLXFERQHONN-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C18H28N2O/c1-4-5-12-20-13-7-6-11-16(20)18(21)19-17-14(2)9-8-10-15(17)3/h8-10,16H,4-7,11-13H2,1-3H3,(H,19,21)
|
| 化学名 |
1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0780 mL | 15.3898 mL | 30.7796 mL | |
| 5 mM | 0.6156 mL | 3.0780 mL | 6.1559 mL | |
| 10 mM | 0.3078 mL | 1.5390 mL | 3.0780 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Efficacy and Safety of Liposomal Bupivacaine Injection for Paravertebral Nerve Block in the Treatment of Acute and Chronic Pain After Thoracoscopic Pneumonectomy: a Multicenter, Randomized, Double-blind, Controlled Clinical Trial
CTID: NCT06569953
Phase: Phase 4 Status: Recruiting
Date: 2024-09-26
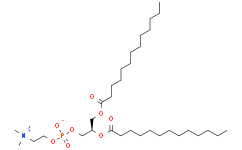 1,2-ditridecanoyl-sn-glycero-3-phosphocholine
1,2-ditridecanoyl-sn-glycero-3-phosphocholine
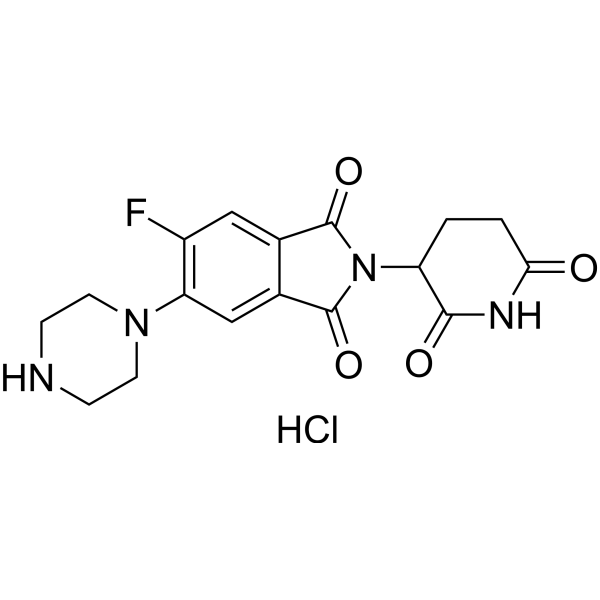 2-(2,6-DIOXOPIPERIDIN-3-YL)-5-FLUORO-6-(PIPERAZIN-1-YL)ISOINDOLINE-1,3-DIONE HCL
2-(2,6-DIOXOPIPERIDIN-3-YL)-5-FLUORO-6-(PIPERAZIN-1-YL)ISOINDOLINE-1,3-DIONE HCL
 2-氯甲基-3-甲基-4-甲氧基吡啶盐酸盐
2-氯甲基-3-甲基-4-甲氧基吡啶盐酸盐
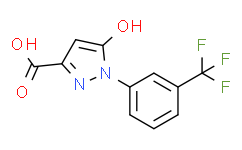 1H-Pyrazole-3-carboxylic acid, 5-hydroxy-1-[3-(trifluoromethyl)phenyl]-
1H-Pyrazole-3-carboxylic acid, 5-hydroxy-1-[3-(trifluoromethyl)phenyl]-
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























