| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|

| 靶点 |
human LPA1 (Kb = 6.9 nM), mouse LPA (Kb = 4.0 nM)[1]
|
|---|---|
| 体外研究 (In Vitro) |
在过表达 LPA1 的 CHO 细胞中,BMS-986278 是一种高亲和力 LPA1 拮抗剂,对人类 LPA1 的 Kb 为 6.9 nM,对小鼠 LPA1 的 Kb 为 4.0 nM [1]。 BMS-986278 的 Kb 为 5.8 nM,在溶血磷脂酸 (LPA) 刺激下可抑制正常人肺成纤维细胞中的钙通量 [1]。
|
| 体内研究 (In Vivo) |
BMS-986278(0.1-10 mg/kg;单次口服剂量)以浓度依赖性方式完全抑制 CD1 小鼠中 LPA 刺激的全身组胺释放 [1]。 BMS-986278(3-30 mg/kg;每天口服两次,持续 14 天)可减少博莱霉素诱导的大鼠胶原沉积/肺纤维化 [1]。 BMS-986278 在临床前物种中的药代动力学 [1] 血浆清除率 ((mL/min)/kg) Vss (L/kg) 口服生物利用度 (%) T1/2 (h) 血浆蛋白结合(% 游离) 小鼠 37 5.5 70 2.5 31.4 大鼠 15 3.5 100 4.5 12.6 猴 2.0 1.6 79 11 0.8
|
| 酶活实验 |
In Vitro Biological Assays/体外生物检测[1]
化合物作为LPA1抑制剂的有效性可以在LPA1功能拮抗剂测定中确定,如下所示: 将过表达人LPA1、小鼠LPA1受体或正常人肺成纤维细胞(NHLF,Lonza,MD,USA)的CHO细胞分别以每孔20000或10000个细胞的体积接种在聚-d-赖氨酸涂覆的384孔微孔板中,并在37°下用5%的CO2孵育过夜。第二天,在37°C下用钙指示剂染料加载细胞30分钟,然后在测定前平衡至RT 30分钟。使用Labcyte Echo声学分配器将溶解并连续稀释在100%DMSO中的测试化合物转移到384孔非结合性表面板上,并用测定缓冲液(1×HBSS(Hanks平衡盐溶液)与钙/镁、20 mM HEPES(4-(2-羟乙基)-1-哌嗪乙磺酸)缓冲液和0.1%无脂肪酸BSA)进一步稀释至0.5%DMSO,然后以0.08 nM至5μM的终浓度加入细胞。随后在室温下孵育细胞20分钟,然后用终浓度为10nM的LPA刺激(EC50)。在加入LPA后,监测LPA刺激的钙通量的抑制90秒。使用四参数逻辑斯谛方程确定IC50值(定义为抑制50%LPA反应的受试化合物的浓度)。使用功能Cheng-Prusoff方程从IC50值计算功能亲和力Kb: 其中[A]是拮抗剂测定中使用的LPA浓度,EC50是导致LPA激动剂控制曲线50%最大活性的LPA的浓度,h是从LPA激动药数据的拟合中获得的斜率值。 体外代谢产物阐明[1] 化合物33(10μM)与小鼠和人类的肝微粒体(1 mg/mL)在NADPH(1 mM)和GSH(1 mM。 在人、猴、狗、大鼠和小鼠的肝细胞中评估了33的代谢。将小鼠、大鼠、狗、猴子和人类的冷冻肝细胞悬浮在Krebs-Henseleit缓冲液(约1×106个细胞/mL)中,在37°C的加湿CO2(5%)培养箱中用33(10μM)培养2小时。反应后,用上述相同的方法处理样品。以相同的方式评估25的代谢。 |
| 细胞实验 |
血清蛋白结合试验[1]
化合物33在一组多物种血清蛋白结合试验中进行了测试,以确定该化合物与各种物种血清蛋白的结合程度。在一次测试中,通过与单个感兴趣物种(人类、大鼠、小鼠或食蟹猴血清)的血清结合,以达到10μM的最终浓度,对33个独立样本进行了三次检测。使用赛默飞世尔公司的双室快速平衡透析(RED)分析板,在37°C下,在10%的二氧化碳气氛中,在磷酸钠缓冲液中进行5小时的透析。在时间零点(T0[血清]和T0[缓冲液])和插管后5小时(平衡后,T5 R h[血清]、T5̷h])收集缓冲液室和血清室的检测样品。通过液相色谱法分析样品,然后进行串联质谱法(LC-MS/MS)分析,以评估透析装置中缓冲液室和血清室之间自由扩散和平衡的化合物分数(百分比)。在LC-MS/MS分析之前,用缓冲液或血清稀释测定样品,以使每个样品中的最终血清浓度相同。随后,在含有分析内标的乙腈中通过蛋白质沉淀提取这些样品。通过LC-MS/MS分析样品,并通过计算每个样品中33与内标的峰面积比来确定33的相对量。结果表示为孵育后33个游离(未结合)百分比、结合百分比和恢复百分比。以类似的方式获得了其他化合物的血清蛋白结合结果。 |
| 动物实验 |
Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat (10 weeks) given bleomycin [1]
Doses: 3, 10 and 30 mg/kg Doses: Orally, twice (two times) daily for 14 days Experimental Results: The percentage of lung section fibrosis area was Dramatically diminished in the 3 mg/kg (48%) and 10 mg/kg (56%) dose groups. LPA Challenge with Plasma Histamine Evaluation (Acute Pharmacodynamic Assay)[1] The effect of compounds on LPA-stimulated histamine release was assessed in 10 week old female (Hsd:ICR) CD-1 mice. (9c,19) Compounds were administered orally (PO) in a vehicle containing 40% PEG400, 10% cremophor, and 50% phosphate buffered saline (PBS) at a dose volume of 10 mL/kg. Two hours after dosing, animals (each dose group = 10 mice) were challenged with a single tail vein injection of PBS or PBS plus 300 μg of lysophosphatidic acid (LPA, 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate [sodium salt]). LPA was prepared in 0.1% fatty acid free BSA/PBS at 2 μg/μL. Two minutes after the LPA injection, mice were sacrificed via decapitation and blood was collected in ethylenediaminetetraacetic acid (EDTA)-coated tubes, spun down (800× gravity, 10 min). The plasma was separated and stored frozen at −80 °C until assayed for compound concentrations and histamine levels. Histamine levels were determined by a competitive enzyme-linked immunosorbent assay (ELISA) as per manufacturer’s instruction (Histamine EIA, Oxford Biomedical Research). The percentage histamine release was calculated by designating the mean of the PBS plus the LPA group as 100% histamine release, and the PBS group minus LPA as 0% histamine release. [1] Rat Bleomycin Chronic Efficacy Studies[1] Compounds were assessed in a 21 day bleomycin-induced lung fibrosis model modified from the literature. (9c) Male Sprague-Dawley rats, aged 10 weeks, were purchased from Charles River Laboratories. After a 10 day acclimation, rats were anesthetized with isoflurane (4% in 100% O2) and administered bleomycin (3.5 U/kg, delivered in a volume of 2 μL/g sterile saline) via oropharyngeal instillation, using a method modified from the literature. (20a) Rats were subsequently returned to their cages until they fully recovered from anesthesia and were monitored daily for the duration of the experiment. On day 7 post-bleomycin instillation, animals were randomized into groups of 10–12, on the basis of their body weight loss since instillation, and compound dosing was initiated on day 8. On day 21, rats were euthanized, left lung lobes were removed, inflated, fixed in formalin, and processed into paraffin blocks, which were sectioned and stained with Picrosirius Red (PSR) (20c) or Masson’s trichrome (MT). (20d) Stained slides were digitized and were analyzed via HALO software a semiautomated histological scoring system. PSR staining was scored by analyzing the entire lung section for the proportion of tissue stained red (%PSR positive), indicating the presence of collagen. MT stained slides were analyzed for areas of increased optical density (%fibrotic area), as a surrogate for the proportion of lung displaying bleomycin-induced injury/inflammation. [1] Mouse Pharmacokinetic Studies[1] The mouse pharmacokinetics study of BMS-986278 (compound 33) was carried out in male CD-1 mice. Animals (N = 4) were fasted overnight (for oral administration only) and received 33 either as an IV bolus dose (1 mg/kg) via a tail vein or by oral gavage (3 mg/kg). Blood samples (∼0.2 mL) were obtained by retro-orbital bleeding at 0.05 (or 0.25 for oral), 0.5, 1, 4, 7, and 24 h postdose. Within each group, half of the animals were bled at 0.05 (or 0.25 for oral), 0.5, 1, 3, 5, 7, and 24 h. Blood samples were allowed to coagulate and centrifuged at 4 °C (1500–2000 × gravity) to obtain serum. Serum samples were stored at −20 °C until analysis by LC-MS/MS (Characterization of 25). Compound 33 was formulated in 5% dimethyl acetamide and 95% tris buffer for IV administration. For PO administration at 3 mg/kg, 33 was formulated as a solution in 10% cremophor-EL, 40% PEG400, and 50% aqueous phosphate buffer. Blood samples were collected; serum was prepared and stored as described above until analysis by LC-MS/MS. A similar protocol was used for the mouse PK study of 25.[1] Rat Pharmacokinetics Studies[1] The rat pharmacokinetics study of BMS-986278 (compound 33) was carried out in male Sprague-Dawley rats (300–350 g). Rats were fasted overnight for PO administration only. Blood samples (∼0.3 mL) were collected from the jugular vein into tripotassium ethylenediaminetetraacetic acid (K3EDTA)-containing tubes at 0.17 (IV only), 0.25, 0.5, 0.75, 1, 2, 3, 5, 7, and 24 h postdose and then centrifuged at 4 °C (1500–2000g) to obtain plasma, which was stored at −20 °C until analysis by LC-MS/MS (Characterization of 25). A similar protocol was used in the rat PK studies of 25 and the other compounds for which rat PK data are described in this paper.[1] Cynomolgus Monkey Pharmacokinetic Studies[1] The monkey pharmacokinetics profile of BMS-986278 (compound 33) was evaluated in a crossover-design study in male cynomolgus monkeys. Following an overnight fast, 3 animals (5–8 kg) received 33 by IV infusion (1 mg/kg over 10 min) via a femoral vein and by oral gavage (5 mg/kg), with a 1 week washout between treatments. Serial blood samples (∼0.3 mL) were collected from a femoral artery into K3EDTA-containing tubes predose and at 0.17 (IV only), 0.25, 0.5, 0.75, 1, 2, 3, 5, 7, and 24 h postdose and centrifuged at 4 °C (1500–2000g) to obtain plasma. Samples were stored at −20 °C until analysis by LC-MS/MS (Characterization of 25). A similar protocol was followed for the cynomolgus monkey PK study of 25. |
| 参考文献 | |
| 其他信息 |
The oxycyclohexyl acid BMS-986278 (33) is a potent lysophosphatidic acid receptor 1 (LPA1) antagonist, with a human LPA1 Kb of 6.9 nM. The structure-activity relationship (SAR) studies starting from the LPA1 antagonist clinical compound BMS-986020 (1), which culminated in the discovery of 33, are discussed. The detailed in vitro and in vivo preclinical pharmacology profiles of 33, as well as its pharmacokinetics/metabolism profile, are described. On the basis of its in vivo efficacy in rodent chronic lung fibrosis models and excellent overall ADME (absorption, distribution, metabolism, excretion) properties in multiple preclinical species, 33 was advanced into clinical trials, including an ongoing Phase 2 clinical trial in patients with lung fibrosis (NCT04308681).[1]
Compound 33 is a potent LPA1 antagonist in vitro (LPA1Kb = 6.9 nM in CHO cells overexpressing human LPA1 and LPA1Kb = 5.9 nM in normal human lung fibroblasts). In a 21 day study (in therapeutic mode) in the chronic rat bleomycin lung fibrosis model, 33 demonstrated robust antifibrotic efficacy; it significantly decreased lung fibrosis area in the bleomycin-treated rats at the 3 and 10 mg/kg BID doses, as determined by histological analysis (Masson’s trichrome staining) at study termination. Overall, 33 has an excellent in vitro liability profile, with (1) no inhibition of any key CYP450 enzymes or ion channels (hERG, sodium, and L-type calcium) that would represent safety concerns and (2) no induction of CYP450 enzymes in human hepatocytes. Importantly, 33 has negligible activity at key hepatic transporters, especially the hepatobiliary bile acid transporters such as BSEP, MDR3, MRP3, and MRP4. An excellent pharmacokinetic profile for 33 was observed in three preclinical species (mouse, rat, and monkey). The discovery of 33 accomplished our objective of significantly improving upon 1 (hepatic/DDI transporter inhibition, aqueous solubility and human hepatocyte metabolic stability), thus validating our design strategy of reducing cLogP and increasing C(sp3) fraction. In conclusion, 33 is a novel LPA1 receptor antagonist with an excellent biological and liability profile which has promise for the treatment of fibrotic diseases. Compound 33 has been advanced into clinical studies; a single-ascending dose and a multiple-ascending dose study have both been completed. A Phase 2 clinical trial with 33 in patients with IPF and PF-ILD has been initiated (ClinicalTrials.gov Identifier NCT04308681).[1] BMS-986278 (33) has to date completed several early stage clinical trials, including a single ascending dose (SAD) study and a 14 day multiple ascending dose (MAD) study in normal healthy volunteers. Compound 33 was generally well tolerated at all doses up to 125 mg BID. Linear and dose-proportional pharmacokinetics was observed for 33 with a very low degree of accumulation following either once daily or twice daily dosing. At the highest dose of 125 mg twice daily, the daily exposure of 33 as defined by AUC was ∼17-fold higher than the AUC achieved with the 600 mg twice daily dose of 1. In terms of pharmacokinetics in normal healthy volunteers, 33 was rapidly absorbed orally with a Tmax of 1.5 h and a half-life of ∼12 h. A global Phase 2 clinical trial in patients with IPF or PF-ILD has been initiated with 33 (ClinicalTrials.gov Identifier NCT04308681).[1] |
| 分子式 |
C22H31N5O5
|
|---|---|
| 分子量 |
445.5120
|
| 精确质量 |
445.23
|
| 元素分析 |
C, 59.31; H, 7.01; N, 15.72; O, 17.96
|
| CAS号 |
2170126-74-4
|
| PubChem CID |
132232205
|
| 外观&性状 |
White to light yellow solid powder
|
| LogP |
2.1
|
| tPSA |
120
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
638
|
| 定义原子立体中心数目 |
2
|
| SMILES |
O(C1C([H])=C([H])C(C2=C(C([H])([H])OC(N(C([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])[H])=O)N(C([H])([H])[H])N=N2)=NC=1C([H])([H])[H])[C@@]1([H])C([H])([H])C([H])([H])C([H])([H])[C@]([H])(C(=O)O[H])C1([H])[H]
|
| InChi Key |
UEUNDURNLYLSNB-HOTGVXAUSA-N
|
| InChi Code |
InChI=1S/C22H31N5O5/c1-5-11-26(3)22(30)31-13-18-20(24-25-27(18)4)17-9-10-19(14(2)23-17)32-16-8-6-7-15(12-16)21(28)29/h9-10,15-16H,5-8,11-13H2,1-4H3,(H,28,29)/t15-,16-/m0/s1
|
| 化学名 |
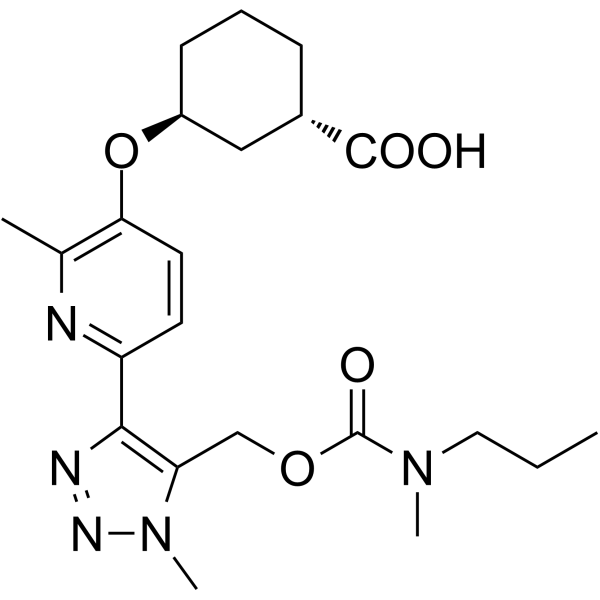
(1S,3S)-3-[2-methyl-6-[1-methyl-5-[[methyl(propyl)carbamoyl]oxymethyl]triazol-4-yl]pyridin-3-yl]oxycyclohexane-1-carboxylic acid
|
| 别名 |
BMS-986278; 2170126-74-4; Admilparant; 4UN9AOU6G8; UNII-4UN9AOU6G8; BMS986278; (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid; Cyclohexanecarboxylic acid, 3-((2-methyl-6-(1-methyl-5-((((methylpropylamino)carbonyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)-3-pyridinyl)oxy)-, (1S,3S)-;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~224.46 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.61 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.61 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.61 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2446 mL | 11.2231 mL | 22.4462 mL | |
| 5 mM | 0.4489 mL | 2.2446 mL | 4.4892 mL | |
| 10 mM | 0.2245 mL | 1.1223 mL | 2.2446 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























