
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
11β-HSD1/11β-hydroxysteroid dehydrogenase type 1
|
|---|---|
| 体外研究 (In Vitro) |
除了在治疗 II 型糖尿病方面可能的临床应用外,11β-HSD1 抑制也可能有益于预防动脉粥样硬化和认知能力下降。在 HEK 和 3T3L1 细胞中,BMS-816336 (6n-2) 抑制 11β-HSD1 酶,IC50 值分别为 37.3 和 28.6 nM [1]。
|
| 体内研究 (In Vivo) |
BMS-816336 是一种可能的新型药物,用于治疗代谢综合征、2 型糖尿病和其他糖皮质激素调节的人类疾病。在 DIO 小鼠(1、3、10、30、100 mg/kg,120 分钟)和食蟹猴(ED50=0.12 mg/kg)中,BMS-816336 (6n-2) 显示出强烈的急性药效作用。在临床前物种中,其口服生物利用度为 20% 至 72%。其在人体中的预期药代动力学特性包括半衰期短和峰谷比[1]。
|
| 酶活实验 |
11β-HSD1 SPA酶分析[1]
采用闪烁接近法(SPA)在384孔PerkinElmer白板上检测11β-HSD1。化合物的剂量反应是用11个半对数稀释的化合物在二甲基亚砜中确定的。每孔加入0.5 μL的DMSO复合稀释液。然后加入空白缓冲液15 μL或人微粒体缓冲液15 μL,室温孵育10 min。最终微粒体蛋白浓度为1.1 μg/次。复制品在同一板上一排下一排。每孔加入3h -可的松10 μL(终浓度40 nM),旋下板搅拌,将含量物降至孔底。室温下轻摇培养4 h,加入10 μL的10 mM卡贝诺酮停止反应。将硅酸钇SPA微球偶联抗皮质醇抗体(20 μL) 0.5 mg加入板孔中,再次纺丝,室温孵育过夜。在TopCount中读取板(1分钟/孔)。数据自动上传到工具集,这是一个用于数据捕获和计算的领先评估信息学程序。图形是用Curve Master程序生成的。 |
| 动物实验 |
Animal/Disease Models: non-fasting diet-induced obese male mice [1]
Doses: 1, 3, 10, 30, 100 mg/kg Route of Administration: po (po (oral gavage)) 120 minutes Experimental Results: ED50=8.6 mg/kg, plasma EC50 is 0.85 μM model [1]. In Vivo Pharmacodynamic Assessment in Mice[1] Nonfasting diet-induced obese male mice were weighed and separated into groups (n = 6) such that body weights were not statistically different from each other. Animals were bled via the tail for a −60 min time sample and then were dosed orally with vehicle or drug. The vehicle was composed of 0.5% Methocel, 0.1% Tween 80 in water. At 60 min after dosing, mice were bled again via the tail and dosed orally with DHC @ 10 mg/kg. All animals were subsequently bled at 30, 60, and 120 min post DHC dosing. Plasma was isolated for analysis of corticosterone using a commercially available enzyme immunosorbent assay. Drug levels were also measured in the terminal bleed samples. In Vivo Pharmacodynamic Assessment in Cynomolgus Monkey[1] The experimental design for the cynomolgus monkey pharmacodynamics study is very similar to the mouse model described above. The test compound was administered at a predetermined time point prior to initiation of the experiment based upon the pharmacokinetics of the compound. The monkeys were then given a dose of substrate, and blood samples were removed at various time points thereafter. A notable difference from the mouse protocol was that even though the natural substrate for 11β-HSD-1 in primates was cortisone, the natural rodent substrate 11-dehydrocorticosterone (DHC) was used instead. |
| 药代性质 (ADME/PK) |
Following an IV dose, the total plasma clearance (CLTp) of BMS-816336 (6n-2) was high in rats, moderate in mice and monkeys, and low in dogs. Apparent elimination half-life estimates were 2 h (mouse), 3 h (rat), 7 h (dog), and 6 h (monkey). BMS-816336 (6n-2) distributes extravascularly in all animal species tested, with Vss of 2.0, 0.5, 3.0, and 4.2 L/kg in mouse, rat, dog, and monkey, respectively. In mice, the tissue-to-plasma concentration ratio averaged ∼0.15 in the adipose but showed concentration-dependency in the liver (1 to 56). Compound 6n-2 was deemed not to penetrate the blood–brain barrier in mice as brain concentrations were below the limit of quantification and brain/plasma ratios were found to be <0.1 at all time points studied. The absolute oral bioavailability of 6n-2 given as a homogenized suspension was estimated to be 56% (mouse), 20% (rat), 72% (dog), and 57% (monkey). Overall, 6n-2 demonstrated a favorable in vivo pharmacokinetic profile with higher peak-to-trough ratio, shorter mean residence times (MTR), and higher clearance values, differentiating itself well from our previous two clinical compounds. The pharmacokinetic properties of 6n-2 appeared to be potentially suitable for “transient” inhibition of the enzyme over a 24 h period dosed once daily. As such, we were eager to test this compound for HPA activation in cynomolgus monkey, a study which will be discussed later.[1]
|
| 参考文献 | |
| 其他信息 |
BMS-816336 (6n-2), a hydroxy-substituted adamantyl acetamide, has been identified as a novel, potent inhibitor against human 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzyme (IC50 3.0 nM) with >10000-fold selectivity over human 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2). 6n-2 exhibits a robust acute pharmacodynamic effect in cynomolgus monkeys (ED50 0.12 mg/kg) and in DIO mice. It is orally bioavailable (%F ranges from 20 to 72% in preclinical species) and has a predicted pharmacokinetic profile of a high peak to trough ratio and short half-life in humans. This ADME profile met our selection criteria for once daily administration, targeting robust inhibition of 11β-HSD1 enzyme for the first 12 h period after dosing followed by an "inhibition holiday" so that the potential for hypothalamic-pituitary-adrenal (HPA) axis activation might be mitigated. 6n-2 was found to be well-tolerated in phase 1 clinical studies and represents a potential new treatment for type 2 diabetes, metabolic syndrome, and other human diseases modulated by glucocorticoid control.[1]
In summary, 6n-2 is a potent and highly selective human 11β-HSD1 inhibitor, with excellent aqueous solubility and in vitro and in vivo safety profiles and acceptable pharmacokinetic properties. Structure-based drug design contributed to the incorporation of the 6-hydroxy group in the adamantane ring as the optimal substitution for activity and metabolic stability. Despite structural similarities, we observed flipped binding orientations of 4k and 6n-2 in their cocrystal structures with human 11β-HSD1 enzyme. The PK profile of 6n-2 was selected to enable partial (e.g., 12 h) enzyme inhibition in vivo upon once daily dosing, allowing for a “holiday” from inhibition, potentially resulting in amelioration of HPA axis activation typically observed with other inhibitors of this enzyme. While we suspect the pharmacokinetic profile of 6n-2 may alter its ability to activate the HPA axis, it is also noted that 6n-2 does not reach the brain in significant concentrations (brain:plasma ratio ∼0.05 in preclinical species), and this may also contribute to the lack of apparent HPA axis activation in the cyno. On the basis of an acceptable pre-IND safety and toxicity profile, 6n-2 has been advanced to phase 1 clinical studies and was found to be well-tolerated up to the maximally tested dose of 900 mg. The data from this study will be reported in due course. In addition to the treatment of type II diabetes and metabolic syndrome, 11β-HSD1 inhibition may find other potential clinical utilities such as atheroprotection and cognitive protection. These areas await further exploration.[1] |
| 分子式 |
C21H27NO3
|
|---|---|
| 分子量 |
341.443986177444
|
| 精确质量 |
341.199
|
| 元素分析 |
C, 73.87; H, 7.97; N, 4.10; O, 14.06
|
| CAS号 |
1009583-20-3
|
| 相关CAS号 |
(Rac)-BMS-816336;(R)-BMS-816336;1009583-83-8; 1009583-20-3 1009365-98-3
|
| PubChem CID |
59336911
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
1.7
|
| tPSA |
60.8
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
25
|
| 分子复杂度/Complexity |
505
|
| 定义原子立体中心数目 |
0
|
| SMILES |
OC1C2CC3CC1CC(C2)C3(C1C=CC=CC=1)CC(N1CC(C1)O)=O
|
| InChi Key |
OAAZMUGLOXGVNH-MEMOLBONSA-N, OAAZMUGLOXGVNH-CCVYDFRESA-N
|
| InChi Code |
InChI=1S/C21H27NO3/c23-18-11-22(12-18)19(24)10-21(15-4-2-1-3-5-15)16-6-13-7-17(21)9-14(8-16)20(13)25/h1-5,13-14,16-18,20,23,25H,6-12H2/t13-,14?,16-,17+,20-,21-/m1/s1
|
| 化学名 |
2-((1R,2R,3S,5R,6R)-6-hydroxy-2-phenyladamantan-2-yl)-1-(3-hydroxyazetidin-1-yl)ethan-1-one
|
| 别名 |
BMS-816336; BMS 816336; 1009583-20-3; (Rac)-BMS-816336; (R)-BMS-816336; HLF8J24L87; 1009583-83-8; 1-(3-hydroxyazetidin-1-yl)-2-(6-hydroxy-2-phenyl-2-adamantyl)ethanone; Ethanone, 1-(3-hydroxy-1-azetidinyl)-2-(6-hydroxy-2-phenyltricyclo(3.3.1.13,7)dec-2-yl)-;
BMS816336.
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~300 mg/mL (~878.63 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 7.5 mg/mL (21.97 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 75.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 7.5 mg/mL (21.97 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 75.0mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 7.5 mg/mL (21.97 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9288 mL | 14.6439 mL | 29.2877 mL | |
| 5 mM | 0.5858 mL | 2.9288 mL | 5.8575 mL | |
| 10 mM | 0.2929 mL | 1.4644 mL | 2.9288 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
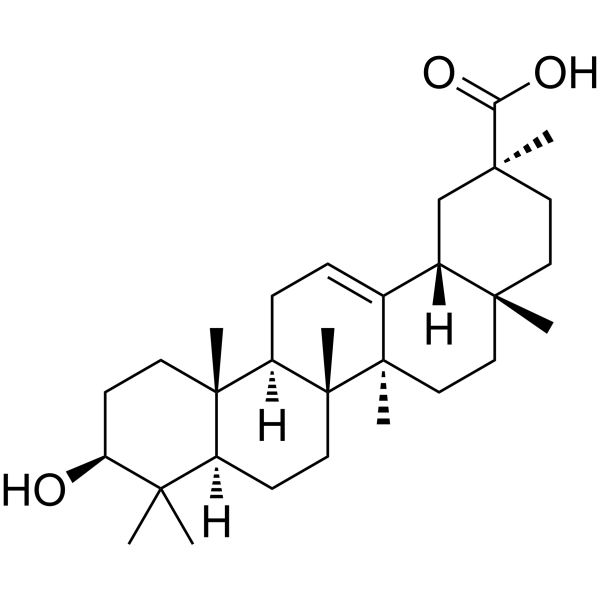 DG 381B
DG 381B
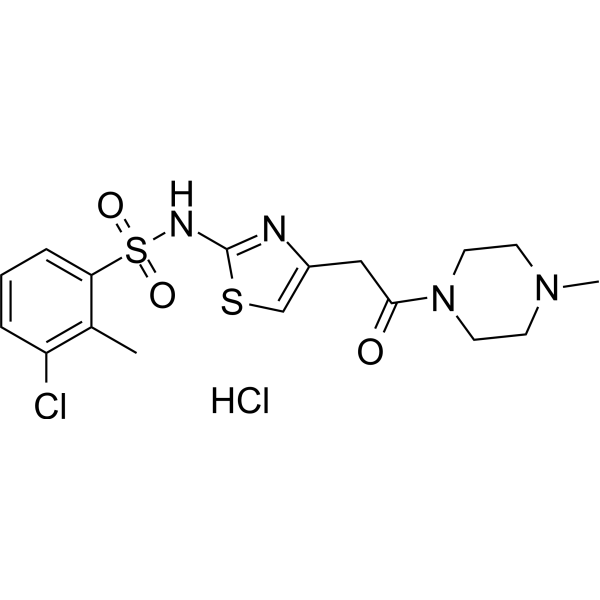 BVT-2733 hydrochloride
BVT-2733 hydrochloride
 UE2343
UE2343
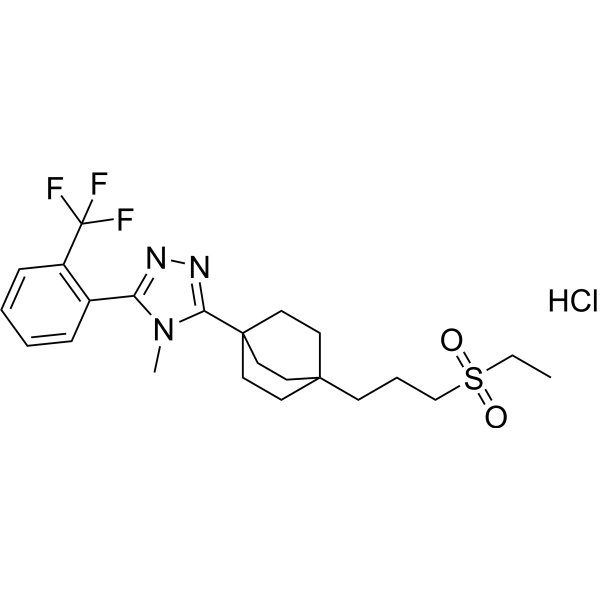 MK-0736 hydrochloride
MK-0736 hydrochloride
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
