| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
PD-1/PD-L1 PPI; PD-L1 protein
|
|---|---|
| 体外研究 (In Vitro) |
其中一个PD-L1单体往往比另一个单体具有更稳定的与BMS-8的结合模式,并且通过Ile54、Tyr56、Met115、Ala121的非极性相互作用进一步稳定诱导PD-L1二聚化的小分子和 Tyr123 位于 ALys124 的单体和水桥上[2]。
|
| 体内研究 (In Vivo) |
NP19 [[an analog of BMS-8]]在H22肝癌小鼠模型[3]
中的体内抗肿瘤活性 鉴于NP19在黑色素瘤B16-F10肿瘤模型中具有良好的体内抗肿瘤效果,且PD-1/PD-L1抑制剂具有广谱的抗肿瘤活性,我们进一步利用H22肝癌模型BALB/c小鼠对复方NP19的体内抗肿瘤效果进行了评价。每只小鼠右侧皮下注射80万个H22细胞。当肿瘤体积达到约100 mm3时,随机选取小鼠,通过腹腔注射NP19或载体溶液治疗14天。如图8所示,NP19在25 mg/kg剂量下具有显著的体内抗肿瘤功效,TGI为76.5%(图8A, 8B, 8C)。此外,NP19没有引起明显的体重减轻(图8D),表明该化合物耐受性良好。 <人力资源> NP19 [an analog of BMS-8]在B16-F10小鼠黑色素瘤模型中的体内抗肿瘤活性[3] 为了确定新合成化合物的体外抗pd -1/PD-L1活性是否可以转化为体内功效,我们在小鼠黑色素瘤B16-F10肿瘤模型上测试了化合物NP19的抗肿瘤活性。选择NP19进行体内疗效研究,是因为与更有效的化合物NP2或同样有效的化合物NP12相比,NP19易于合成且细胞毒性较小(表9)。我们将携带黑色素瘤的BALB/c小鼠分别以载药对照和NP19 (25 mg/kg、50 mg/kg、100 mg/kg)灌胃治疗,每天1次,持续15天。如图6所示,治疗15天后,NP19治疗显著抑制了黑色素瘤的生长。 <人力资源> NP19的体内药代动力学性质[an analog of BMS-8][3] 由于化合物NP19在体外表现出较高的药效,接下来通过静脉注射和口服给药来评估雄性Sprague-Dawley大鼠的药代动力学(PK)谱。表8总结了关键po和静脉给药PK参数。单次静脉给药1 mg/kg化合物NP19后,NP19的半衰期(t1/2)为1.5±0.5 h,清除率(CL)为0.9±0.2 L/h/kg,表观分布体积(Vss)为2.1±0.5 L/kg。NP19口服给药剂量为10 mg/kg时,观察到NP19的口服吸收(Tmax = 0.6±0.2 h)、长半衰期(t1/2 = 10.9±7.7 h)和口服生物利用度(F = 5%)。此外,在大鼠中未观察到明显的不良反应。NP19经口服灌胃后的半衰期(10.9 h)比静脉注射的半衰期(1.5 h)长得多;这可能是由于NP19的高亲脂性(logP = 7.9)或水溶性差。结果,NP19表现出翻转式药代动力学。这种翻转式药代动力学有时会发生在像利巴米胺这样水溶性差的化合物上,由于其水溶性差(7.6 μg/mL),其t1/2 (p.o)/t1/2 (i.v)比为13.5。另一个例子是李建明等人报道的亲脂化合物IAT(一种水溶性为19 μg/mL的抗微管蛋白剂),其t1/2 (p.o.)/t1/2 (i.v.)的比值为~ 5,与NP19 [t1/2 (p.o.)/t1/2 (i.v.) = 7.1]相似。由于化合物NP19的口服生物利用度较低,我们推测需要高剂量才能提供足够的药物浓度以显示抗肿瘤功效。因此,我们进一步研究了化合物NP19的体内活性。 |
| 酶活实验 |
所有结合研究均在 HTRF 测定缓冲液中进行,该缓冲液由 dPBS 组成,并补充有 0.1%(含 v)牛血清白蛋白和 0.05%(v/v)Tween-20。对于 PD-l-Ig/PD-Ll-His 结合测定,将抑制剂与 PD-Ll-His(最终浓度为 10 nM)在 4 μL 测定缓冲液中预孵育 15 m,然后添加 PD-l- Ig(最终 20 nM)溶于 1 μL 测定缓冲液中,并进一步孵育 15 m。使用来自人类、食蟹猴或小鼠的 PD-L1。 HTRF 检测是使用铕穴酸盐标记的抗 Ig(最终 1 nM)和别藻蓝蛋白 (APC) 标记的抗 His(最终 20 nM)实现的。将抗体在 HTRF 检测缓冲液中稀释,并在结合反应上分配 5 μL。让反应混合物平衡 30 分钟,并使用 En Vision 荧光计获得信号(665 nm/620 nm 比率)。在 PD-1-Ig/PD-L2-His(分别为 20、5 nM)、CD80-His/PD-Ll-Ig(分别为 100、10 nM)和 CD80-His/CTLA4- 之间建立了额外的结合测定。 Ig(分别为 10、5 nM)。
|
| 细胞实验 |
特别是在肿瘤细胞裂解成为关注焦点的肿瘤微环境中,程序性死亡 1/程序性死亡配体 1 (PD-1/PD-L1) 之间的相互作用在抑制 T 细胞反应中发挥着主导作用。 PD-1/PD-L1 抑制剂 2 的 IC50 值为 18 nM,据说可以阻止 PD-L1 与 PD-1 相互作用。
|
| 动物实验 |
Pharmacokinetic Study in Male Sprague–Dawley Rats[3]
Male Sprague–Dawley rats (200–220 g) were used to study the pharmacokinetics of compound NP19 [an analog of BMS-8]. Diet was prohibited for 12 h before the experiment, but water was freely available. Blood samples (0.3 mL) were collected from the tail vein into heparinized 1.5 mL polythene tubes at 0.0833, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h after oral (10 mg/kg) or intravenous (1 mg/kg) administration of compound NP19. The compound was dissolved in 5% DMSO and 95% PEG-300 for intravenous administration or suspended in 0.5% sodium carboxymethyl cellulose (CMC-Na) for oral administration. The samples were immediately centrifuged at 3000g for 10 min. The plasma as-obtained (100 μL) was stored at −20 °C until analysis. PK parameters were determined from individual animal data using noncompartmental analysis in DAS (Drug and statistics) software. Instruments and analytical conditions for PK studies: A UPLC-MS/MS system with ACQUITY I-Class UPLC and a XEVO TQD triple quadrupole mass spectrometer, equipped with an electrospray ionization (ESI) interface, was used to analyze the blood samples. The UPLC system was comprised of a Binary Solvent Manager (BSM) and a Sample Manager with Flow-Through Needle (SM-FTN). Masslynx 4.1 software was used for data acquisition and instrument control. Multiple reaction monitoring (MRM) modes of m/z 555.35 → 181.03 for NP19 and m/z 237 → 194.1 for carbamazepine were utilized to conduct quantitative analysis. In Vivo Efficacy Study in Mouse B16F10 Melanoma Model[3] BALB/c mice, aged 6–8 weeks old, were used to study the inhibition effect of NP19 [an analog of BMS-8]on subcutaneous transplanted model of melanoma cells. Murine B16F10 melanoma cells growing in a logarithmic growth phase were suspended in PBS at a density of 2 × 106 per mL. Each mouse was inoculated subcutaneously with 200 μL containing 4 × 105 cells. After tumors reached approximately 100 mm3 in volume, mice were divided into four groups randomly (n = 10) and treated with NP19 (25, 50, 100 mg/kg) and vehicle, respectively. The drugs were administered via intragastric gavage once a day for 15 days. The vehicle group was administered with 0.5% sodium carboxymethyl cellulose (CMC-Na). Animal activity and body weight were monitored during the entire experiment period to assess acute toxicity. Mice were sacrificed 16 days after the initiation of the treatment, and the tumor tissue and major organ (liver, spleen, thymus, and kidney) samples were collected. The harvested tumor tissue and organs (liver, kidney) were fixed in 4% paraformaldehyde, processed into paraffin routinely, stained with hematoxylin and eosin (H&E), and captured by microscope. Tumor growth inhibition value (TGI) was calculated using the formula: TGI(%) = [1 – Wt/Wv] × 100%, where Wt and Wv are the mean tumor weight of treatment group and vehicle control. In Vivo Efficacy Study in Mouse H22 Hepatoma Tumor Model[3] 6–8 weeks old male BALB/c mice were used. A total of 8 × 105 H22 cells were inoculated into the right flank of each mouse according to protocols of tumor transplant research. NP19 [an analog of BMS-8]was dissolved in 5% DMSO, 40% PEG-200 and 55% saline solution to produce desired concentrations. Mice in control groups were injected intraperitoneally with 200 μL of vehicle solution only. Tumor volume was measured every 2 days with a traceable electronic digital caliper and calculated using the formula a × b2 × 0.5, where a and b represented the larger and smaller diameters, respectively. The mice were sacrificed after the treatments and tumors were excised and weighed. |
| 参考文献 |
|
| 其他信息 |
Recently, small-molecule compounds have been reported to block the PD-1/PD-L1 interaction by inducing the dimerization of PD-L1. All these inhibitors had a common scaffold and interacted with the cavity formed by two PD-L1 monomers. This special interactive mode provided clues for the structure-based drug design, however, also showed limitations for the discovery of small-molecule inhibitors with new scaffolds. In this study, we revealed the structure-activity relationship of the current small-molecule inhibitors targeting dimerization of PD-L1 by predicting their binding and unbinding mechanism via conventional molecular dynamics and metadynamics simulation. During the binding process, the representative inhibitors (BMS-8 and BMS-1166) tended to have a more stable binding mode with one PD-L1 monomer than the other and the small-molecule inducing PD-L1 dimerization was further stabilized by the non-polar interaction of Ile54, Tyr56, Met115, Ala121, and Tyr123 on both monomers and the water bridges involved in ALys124. The unbinding process prediction showed that the PD-L1 dimerization kept stable upon the dissociation of ligands. It's indicated that the formation and stability of the small-molecule inducing PD-L1 dimerization was the key factor for the inhibitory activities of these ligands. The contact analysis, R-group based quantitative structure-activity relationship (QSAR) analysis and molecular docking further suggested that each attachment point on the core scaffold of ligands had a specific preference for pharmacophore elements when improving the inhibitory activities by structural modifications. Taken together, the results in this study could guide the structural optimization and the further discovery of novel small-molecule inhibitors targeting PD-L1.[2]
Cancer immunotherapy has been revolutionized by the development of monoclonal antibodies (mAbs) that inhibit interactions between immune checkpoint molecules, such as programmed cell-death 1 (PD-1), and its ligand PD-L1. However, mAb-based drugs have some drawbacks, including poor tumor penetration and high production costs, which could potentially be overcome by small molecule drugs. BMS-8, one of the potent small molecule drugs, induces homodimerization of PD-L1, thereby inhibiting its binding to PD-1. Our assay system revealed that BMS-8 inhibited the PD-1/PD-L1 interaction with IC50 of 7.2 μM. To improve the IC50 value, we designed and synthesized a small molecule based on the molecular structure of BMS-8 by in silico simulation. As a result, we successfully prepared a biphenyl-conjugated bromotyrosine (X) with IC50 of 1.5 μM, which was about five times improved from BMS-8. We further prepared amino acid conjugates of X (amino-X), to elucidate a correlation between the docking modes of the amino-Xs and IC50 values. The results suggested that the displacement of amino-Xs from the BMS-8 in the pocket of PD-L1 homodimer correlated with IC50 values. This observation provides us a further insight how to derivatize X for better inhibitory effect.[1] This work describes the modification of a gold electrode with the BMS-8 compound that interacts with the Programmed Death-Ligand 1 (PD-L1), an immune checkpoint protein. The results show that we can confirm the presence of the sPD-L1 in the concentration range of 10-18 to 10-8 M using electrochemical impedance spectroscopy (EIS) with a limit of detection (LOD) of 1.87 × 10-14 M for PD-L1 (S/N = 3.3) and at a concentration of 10-14 M via cyclic voltammetry (CV). Additionally, high-resolution X-ray photoelectron spectroscopy (XPS), contact angle, and surface free energy measurements were applied to confirm the functionalization of the electrode. We investigated the selectivity of the electrode for other proteins: Programmed Death-1 (PD-1), cluster of differentiation 160 (CD160), and B- and T-lymphocyte attenuator (BTLA) at concentrations of 10-8 M. Differentiation between PD-L1 and PD-1 was achieved based on the analysis of the capacitance effect frequency dispersion at the surface of the modified Au electrode with BMS-8 after incubation at various concentrations of PD-L1 and PD-1 proteins in the range of 10-18 to 10-8 M. Significant differences were observed in the heterogeneity of PD-L1 and PD-1. The results of the quasi-capacitance studies demonstrate that BMS-8 strongly and specifically interacts with the PD-L1 protein. https://pubmed.ncbi.nlm.nih.gov/33517203/ |
| 分子式 |
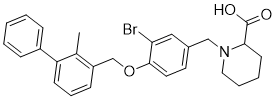
C27H28BRNO3
|
|
|---|---|---|
| 分子量 |
494.43
|
|
| 精确质量 |
493.125
|
|
| 元素分析 |
C, 65.59; H, 5.71; Br, 16.16; N, 2.83; O, 9.71
|
|
| CAS号 |
1675201-90-7
|
|
| 相关CAS号 |
|
|
| PubChem CID |
117941742
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
616.9±55.0 °C at 760 mmHg
|
|
| 闪点 |
326.9±31.5 °C
|
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
|
| 折射率 |
1.620
|
|
| LogP |
6.28
|
|
| tPSA |
49.8
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
32
|
|
| 分子复杂度/Complexity |
596
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
BrC1=C(C=CC(=C1)CN1CCCCC1C(=O)O)OCC1C=CC=C(C2C=CC=CC=2)C=1C
|
|
| InChi Key |
QRXBPPWUGITQLE-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C27H28BrNO3/c1-19-22(10-7-11-23(19)21-8-3-2-4-9-21)18-32-26-14-13-20(16-24(26)28)17-29-15-6-5-12-25(29)27(30)31/h2-4,7-11,13-14,16,25H,5-6,12,15,17-18H2,1H3,(H,30,31)
|
|
| 化学名 |
1-[[3-bromo-4-[(2-methyl-3-phenylphenyl)methoxy]phenyl]methyl]piperidine-2-carboxylic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0225 mL | 10.1127 mL | 20.2253 mL | |
| 5 mM | 0.4045 mL | 2.0225 mL | 4.0451 mL | |
| 10 mM | 0.2023 mL | 1.0113 mL | 2.0225 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2.Structure.2017 Aug 1;25(8):1163-1174. |
|---|
New Directions in Designing the Therapeutics Targeting the PD-1/PD-L1 Interaction.Structure.2017 Aug 1;25(8):1163-1174. |
Structural Basis of the PD-1/PD-L1 (PD-L2) Interaction.Structure.2017 Aug 1;25(8):1163-1174. |
 J Med Chem.2017Jul 13;60(13):5857-5867. |
|---|
|
|
 Cemavafusp
Cemavafusp
 PD-L1-IN-7
PD-L1-IN-7
 CSN5-IN-1
CSN5-IN-1
 PD-1/PD-L1-IN-50
PD-1/PD-L1-IN-50
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA





