| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
Beta-1 adrenergic receptor
|
|---|---|
| 体外研究 (In Vitro) |
比索洛尔(2 μM,1 小时)可保护心肌细胞 (H9c2) 免受缺血/再灌注 (I/R) 损伤[2]。 Bisoprolol(2 μM,1 小时)可减少 H9c2 细胞中 H/R 诱导的 ROS 产生和细胞凋亡[2]。比索洛尔(2 μM,1 小时)可增加 H9c2 细胞中 AKT 和 GSK3β 的磷酸化[2]。 Bisoprolol(100 μM,24 小时)通过增加 β-arrestin 2、CCR7 和 PI3K 磷酸化来逆转胆固醇负载 DC(树突状细胞)中肾上腺素抑制的迁移[3]。细胞活力测定[2] 细胞系:H9c2 细胞 浓度:0.2、2、20 μM 孵育时间:1 h 结果:H/R(缺氧/复氧)心肌细胞存活率提高至 73.20%、90.38%、81.25 % 分别。细胞迁移测定 [3] 细胞系:DC 浓度:100 μM 孵育时间:6、12、24 小时 结果:迁移细胞数量增加 46.00%(6 小时)、64.25%(12 小时)和 55.74%(24 H)。
|
| 体内研究 (In Vivo) |
比索洛尔(口服,5 mg/kg,持续1周)可增加左心室射血分数(LVEF)并降低心率值[2]。比索洛尔(口服灌胃,8 mg/kg,每天,持续四周)对大鼠镉诱导的心肌毒性具有保护作用[4]。比索洛尔(口服灌胃,1 mg/kg,每天,持续 6 周)可逆转容量超负荷大鼠模型中的小电导钙激活钾通道 (SK) 重塑[5]。动物模型:缺血/再灌注(I/R)损伤大鼠[2] 剂量:0.5、5、10 mg/kg 给药方法:口服给药,持续1周,在0.5小时缺血/4小时再灌注之前。结果:梗死面积从 I/R 组的 44% 减少到治疗组的 31%。动物模型:镉诱导大鼠[4] 剂量:2、8 mg/kg 给药方法:口服灌胃,每天一次,持续四个星期。结果:平均动脉压 (MAP) 降低至 8 mg/kg。 8 mg/kg 时血清生物标志物(ALT、AST)和 NF-kB p65 表达以及 TNF-α 水平(心脏组织样本)降低。
|
| 细胞实验 |
细胞系:H9c2细胞
浓度:0.2、2、20 μM 孵育时间:1 h 结果:H/R(缺氧/复氧)心肌细胞存活率提高至73.20%、90.38 %、81.25%。 |
| 动物实验 |
Ischemia/reperfusion (I/R) injury rats
0.5, 5, 10 mg/kg Oral administration, for 1 week, prior to 0.5 h ischemia/4 h reperfusion. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Bisoprolol is well absorbed in the gastrointestinal tract. The AUC is 642.87 g.hr/mL and bioavailability of bisoprolol is about 90% due to the minimal first pass effects. Absorption is unaffected by food intake. Peak plasma concentrations of bisoprolol are attained within 2-4 hours and steady-state concentrations are achieved within 5 days of administration. In a pharmacokinetic study, the mean peak concentration of bisoprolol was 52 micrograms/L. Cmax at steady state concentrations of bisoprolol is 64±21 ng/ml administered at 10 mg daily. Bisoprolol is eliminated equally by both renal and hepatic pathways. About 50% of an oral dose is excreted unchanged in the urine with the remainder of the dose excreted as inactive bisoprolol metabolites. Under 2% of the ingested dose is found to be excreted in the feces. The volume of distribution of bisoprolol is 3.5 L/kg. The mean volume of distribution was found to be 230 L/kg in heart failure patients, which was similar to the volume of distribution in healthy patients. Bisoprolol is known to cross the placenta. Total body clearance in healthy patients was determined to be 14.2 L/h. In patients with renal impairment, clearance was reduced to 7.8 L/h. Hepatic dysfunction also reduced the clearance of bisoprolol. Beagles were treated with bisoprolol, a beta 1-selective adrenoceptor antagonist, for 30 days with the following daily doses: oral: 30 mg/kg; conjunctival: 0.5% solution (approx. 0.04 mg/kg) and 5% solution (approx. 0.4 mg/kg). Drug concentrations were determined in plasma and various eye tissues on days 1, 16, and 30, and on day 59, i.e. on day 29 of the follow-up period. Bisoprolol concentrations in plasma and most eye tissues were considerably higher after oral than after conjunctival treatment. The highest tissue concentrations were observed in the iris (+ciliary body) and retina (+choroid) with tissue/plasma concentration ratios between 100 and 150 after oral and 1000 to 3000 after conjunctival instillation (5% solution). In plasma no accumulation of the drug was observed which is in accordance with its plasma half-life of 4 to 5 hr. In contrast to this, concentrations in the iris and retina increased from day 1 to day 16 and 30 by 3 to 8 times and the half-life of bisoprolol in these tissues was estimated to be between 3 to 5 days. The pharmacokinetic properties of bisoprolol-(14)C were studied in Wistar rats, beagle dogs, and Cynomolgus monkeys. Bisoprolol is well absorbed in these species; independent of the route of administration (IV or PO), 70-90% of the (14)C-dose was recovered in urine. Fecal excretion was approximately 20% in rats and less than 10% in dogs and monkeys. Rats excreted approximately 10% of the dose in bile after IV as well as after oral administration. The plasma half-life of the unchanged drug was approximately 1 hr in rats, 3 hr in monkeys, and 5 hr in dogs. The bioavailability was 40-50% in monkeys, approximately 80% in dogs, and 10% in rats. Studies in rats have shown that the drug is rapidly taken up by the tissues. After IV administration, high levels of radioactivity were found in lung, kidneys, liver, adrenals, spleen, pancreas, and salivary glands. After oral administration, the highest concentration occurred in the liver and kidneys. With the exception of plasma and liver, unchanged bisoprolol was the major radioactive constituent in all tissues studied. Both the blood-brain and placental barriers were penetrated, but only to a small degree. No accumulation of radioactivity in tissues was observed after repeated dosing (1 mg/kg/day). The metabolism of bisoprolol was studied in the same three animal species and in humans. The major metabolites are the products of O-dealkylation and subsequent oxidation to the corresponding carboxylic acids. The amount of bisoprolol excreted unchanged in the urine is 50-60% of the dose in humans, 30-40% in dogs, and approximately 10% in rats and monkeys. The pharmacokinetics of bisoprolol (I) following an oral dose of 20 mg (14)C-labeled I solution, 10 mg tablet, and intravenous injection of 10 mg I were studied in 23 healthy volunteers (aged 37-53 yr). Mean elimination half-lives of 11 h for the unchanged I and 12 h for total radioactivity were observed. The enteral absorption of I was nearly complete. Fifty percent of the dose was eliminated renally as unchanged I and the other 50% metabolically, with subsequent renal excretion of the metabolites. Less than 2% of the dose was recovered from the feces. Intraindividual comparison of the pharmacokinetic data measured after oral or IV dose yielded an absolute bioavailability of 90%. Total and renal clearance were calculated as 15.6 l/h and 9.6 l/h, respectively. The volume of distribution was 226 l. Concomitant food intake did not influence the bioavailability of I. We previously reported that renal function is partly responsible for the interindividual variability of the pharmacokinetics of bisoprolol. The aim of the present study was to examine the variability of bioavailability (F) of bisoprolol in routinely treated Japanese patients and intestinal absorption characteristics of the drug. We first analyzed the plasma concentration data of bisoprolol in 52 Japanese patients using a nonlinear mixed effects model. We also investigated the cellular uptake of bisoprolol using human intestinal epithelial LS180 cells. The oral clearance (CL/F) of bisoprolol in Japanese patients was positively correlated with the apparent volume of distribution (V/F), implying variable F. The uptake of bisoprolol in LS180 cells was temperature-dependent and saturable, and was significantly decreased in the presence of quinidine and diphenhydramine. In addition, the cellular uptake of bisoprolol dissolved in an acidic buffer was markedly less than that dissolved in a neutral buffer. These findings suggest that the rate/extent of the intestinal absorption of bisoprolol is another cause of the interindividual variability of the pharmacokinetics, and that the uptake of bisoprolol in intestinal epithelial cells is highly pH-dependent and also variable. For more Absorption, Distribution and Excretion (Complete) data for Bisoprolol (9 total), please visit the HSDB record page. Metabolism / Metabolites Approximately 50% of the bisoprolol dose is eliminated by non-renal pathways. Bisoprolol is metabolized through oxidative metabolic pathways with no subsequent conjugation. Bisoprolol metabolites are polar and, therefore, really eliminated. Major metabolites found in plasma and urine are inactive. Bisoprolol is mainly metabolized by CYP3A4 (95%), whereas CYP2D6 plays a minor role. The CYP3A4-mediated metabolism of bisoprolol appears to be non-stereoselective. ... In humans, the known metabolites are labile or have no known pharmacologic activity. ... Bisoprolol fumarate is not metabolized by cytochrome P450 II D6 (debrisoquin hydroxylase). The plasma concentrations and urinary excretions of bisoprolol enantiomers in four Japanese male healthy volunteers after a single oral administration of 20 mg of racemic bisoprolol were evaluated. The AUC(infinity) and elimination half-life of (S)-(-)-bisoprolol were slightly larger than those of (R)-(+)-bisoprolol in all subjects. The metabolic clearance of (R)-(+)-bisoprolol was significantly (P < 0.05) larger than that of (S)-(-)-bisoprolol (S/R ratio: 0.79+/-0.03), although the difference was small. In contrast, no stereoselective in vitro protein binding of bisoprolol in human plasma was found. An in vitro metabolic study using recombinant human cytochrome P450 (CYP) isoforms indicated that oxidation of both bisoprolol enantiomers was catalyzed by the two isoforms, CYP2D6 and CYP3A4. CYP2D6 metabolized bisoprolol stereoselectively (R > S), whereas the metabolism of bisoprolol by CYP3A4 was not stereoselective. The S/R ratio of the mean clearance due to renal tubular secretion was 0.68, indicating a moderate degree of stereoselective renal tubular secretion. These findings taken together suggest that the small differences in the pharmacokinetics between (S)-(-)- and (R)-(+)-bisoprolol are mainly due to the stereoselectivity in the intrinsic metabolic clearance by CYP2D6 and renal tubular secretion. The pharmacokinetic properties of bisoprolol-(14)C were studied in Wistar rats, beagle dogs, and Cynomolgus monkeys. ... The metabolism of bisoprolol was studied in the same three animal species and in humans. The major metabolites are the products of O-dealkylation and subsequent oxidation to the corresponding carboxylic acids. ... Biological Half-Life A pharmacokinetic study in 12 healthy individuals determined the mean plasma half-life of bisoprolol to be 10-12 hours. Another study comprised of healthy patients determined the elimination half-life to be approximately 10 hours. Renal impairment increased the half-life to 18.5 hours. In patients with cirrhosis of the liver, the elimination of Zebeta (bisoprolol fumarate) is more variable in rate and significantly slower than that in healthy subjects, with plasma half-life ranging from 8.3 to 21.7 hours. In subjects with creatinine clearance less than 40 mL/min, the plasma half-life is increased approximately threefold compared to healthy subjects. The plasma elimination half-life is 9-12 hours and is slightly longer in elderly patients, in part because of decreased renal function in that population. The pharmacokinetic properties of bisoprolol-(14)C were studied in Wistar rats, beagle dogs, and Cynomolgus monkeys. ... The plasma half-life of the unchanged drug was approximately 1 hr in rats, 3 hr in monkeys, and 5 hr in dogs. In dogs, bisoprolol has ... a half life of 4 hours |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Bisoprolol is a beta-Adrenergic blocking agent, sometimes under the drug name Zebeta, which is indicated in the management of hypertension. It may be used alone or in combination with other antihypertensive agents. HUMAN EXPOSURE AND TOXICITY: The most common signs expected with overdosage of a beta-blocker are bradycardia, hypotension, congestive heart failure, bronchospasm, and hypoglycemia. There have been at least two reported cases where a switch from propranolol to bisoprolol resulted in worsening of arrhythmia control. An elderly person died of uncontrolled bradycardia in a hospital after being mistakenly given a higher-than-prescribed dose of bisoprolol fumarate. However, it was determined that the patient had a mutation within cytochrome P2D6, which influences metabolism of the drug. ANIMAL STUDIES: Fetotoxicity in rats occurred at 125 times the maximum recommended human dose (MRHD) of bisoprolol fumarate on a body-weight-basis, and maternal toxicity occurred at 375 times the MRHD. In rabbits, bisoprolol fumarate was not teratogenic at doses up to 12.5 mg/kg/day (31 times the MRHD based on body-weight), but increased early resorptions. The mutagenic potential of bisoprolol fumarate was evaluated in the microbial mutagenicity (Ames) test, the point mutation and chromosome aberration assays in Chinese hamster V79 cells, the unscheduled DNA synthesis test, the micronucleus test in mice, and cytogenetics assay in rats. There was no evidence of mutagenic potential in these in vitro and in vivo assays. Long-term studies were conducted with oral bisoprolol fumarate administered in the feed of mice and rats. No evidence of carcinogenic potential was seen in mice dosed up to 250 mg/kg/day or rats dosed up to 123 mg/kg/day. Hepatotoxicity Bisoprolol therapy has been associated with a low rate of mild-to-moderate elevations of serum aminotransferase levels which are usually asymptomatic and transient and resolve even with continuation of therapy. There have been no well documented cases of clinically apparent, acute liver injury attributable to bisoprolol. Thus, hepatotoxicity due to bisoprolol must be very rare, if it occurs at all. Most commonly used beta-blockers have been linked to rare instances of clinically apparent liver injury, typically with onset within 2 to 12 weeks, a hepatocellular pattern of liver enzyme elevations, rapid recovery upon withdrawal, and little evidence of hypersensitivity (rash, fever, eosinophilia) or autoantibody formation. Likelihood score: E (unlikely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Limited information indicates that a maternal dose of 5 mg daily produces low levels in milk and some follow-up date indicate no adverse long-term effects on the breastfed infant. If bisoprolol is required by the mother, it is not a reason to discontinue breastfeeding. Other beta-blockers with more safety data may be preferred. ◉ Effects in Breastfed Infants A woman was diagnosed with Cushing's disease during pregnancy. Postpartum she took metyrapone 250 mg 3 times daily, bisoprolol 10 mg twice daily, and captopril 12.5 mg twice daily. She breastfed her preterm infant about 50% milk and 50% formula. At 5 weeks postpartum, the infant's pediatric team found his growth and development to be appropriate. A prospective study followed 11 women who were taking bisoprolol in a median dose was 2.5 mg daily (range 1 to 5 mg) during breastfeeding (8 exclusively). The median age of the child at the time of follow-up was 49 (IRQ 25.5to 58.5) months. Adverse effects were reported among 2 infants: 1 with somnolence and 1 with poor weight gain. No abnormal results were found by Denver developmental scale. Median psychomotor development according to PEDsQL score total 97.5, psychosocial health 97.9 and physical health 100, all representing normal development. ◉ Effects on Lactation and Breastmilk A study in 6 patients with hyperprolactinemia and galactorrhea found no changes in serum prolactin levels following beta-adrenergic blockade with propranolol. Relevant published information on the effects of beta-blockade or bisoprolol during normal lactation was not found as of the revision date. Protein Binding Binding to serum proteins is approximately 30%. Interactions Concurrent use of rifampin increases the metabolic clearance of Zebeta, resulting in a shortened elimination half-life of Zebeta. However, initial dose modification is generally not necessary. Pharmacokinetic studies document no clinically relevant interactions with other agents given concomitantly, including thiazide diuretics and cimetidine. There was no effect of Zebeta on prothrombin time in patients on stable doses of warfarin. Both digitalis glycosides and beta-blockers slow atrioventricular conduction and decrease heart rate. Concomitant use /with Zebeta/ can increase the risk of bradycardia. Zebeta should be used with care when myocardial depressants or inhibitors of AV conduction, such as certain calcium antagonists (particularly of the phenylalkylamine (verapamil) and benzothiazepine (diltiazem) classes), or antiarrhythmic agents, such as disopyramide, are used concurrently. Zebeta should not be combined with other beta-blocking agents. Patients receiving catecholamine-depleting drugs, such as reserpine or guanethidine, should be closely monitored, because the added beta-adrenergic blocking action of Zebeta may produce excessive reduction of sympathetic activity. In patients receiving concurrent therapy with clonidine, if therapy is to be discontinued, it is suggested that Zebeta be discontinued for several days before the withdrawal of clonidine. beta-Blockers may exacerbate the rebound hypertension which can follow the withdrawal of clonidine. If the two drugs are coadministered, the beta-blocker should be withdrawn several days before discontinuing clonidine. If replacing clonidine by beta-blocker therapy, the introduction of beta-blockers should be delayed for several days after clonidine administration has stopped. Non-Human Toxicity Values LD50 Dog iv 24 mg/kg LD50 Dog po 90 mg/kg LD50 Rat iv 50 mg/kg LD50 Rat po 1112 mg/kg For more Non-Human Toxicity Values (Complete) data for Bisoprolol (6 total), please visit the HSDB record page. |
| 参考文献 |
|
| 其他信息 |
Bisoprolol is a secondary alcohol and a secondary amine. It has a role as an antihypertensive agent, a beta-adrenergic antagonist, an anti-arrhythmia drug and a sympatholytic agent.
Bisoprolol is a cardioselective β1-adrenergic blocking agent used to treat high blood pressure. It is considered a potent drug with a long-half life that can be used once daily to reduce the need for multiple doses of antihypertensive drugs. Bisoprolol is generally well tolerated, likely due to its β1-adrenergic receptor selectivity and is a useful alternative to non-selective β-blocker drugs in the treatment of hypertension such as [Carvedilol] and [Labetalol]. It may be used alone or in combination with other drugs to manage hypertension and can be useful in patients with chronic obstructive pulmonary disease (COPD) due to its receptor selectivity. Bisoprolol is a beta-Adrenergic Blocker. The mechanism of action of bisoprolol is as an Adrenergic beta-Antagonist. Bisoprolol is a cardioselective beta-blocker used in the treatment of hypertension. Bisoprolol has not been linked to instances of clinically apparent drug induced liver injury. Bisoprolol Fumarate is the fumarate salt of a synthetic phenoxy-2-propanol-derived cardioselective beta-1 adrenergic receptor antagonist with antihypertensive and potential cardioprotective activities. Devoid of intrinsic sympathomimetic activity, bisoprolol selectively and competitively binds to and blocks beta-1 adrenergic receptors in the heart, decreasing cardiac contractility and rate, reducing cardiac output, and lowering blood pressure. In addition, this agent may exhibit antihypertensive activity through the inhibition of renin secretion by juxtaglomerular epithelioid (JGE) cells in the kidney, thus inhibiting activation of the renin-angiotensin system (RAS). Bisoprolol has been shown to be cardioprotective in animal models. Bisoprolol is a selective beta-1 adrenergic receptor antagonist with antihypertensive activity and devoid of intrinsic sympathomimetic activity. Bisoprolol selectively and competitively binds to and blocks beta-1 adrenergic receptors in the heart, thereby decreasing cardiac contractility and rate. This leads to a reduction in cardiac output and lowers blood pressure. In addition, bisoprolol prevent the release of renin, a hormone secreted by the kidneys that causes constriction of blood vessels. A cardioselective beta-1 adrenergic blocker. It is effective in the management of HYPERTENSION and ANGINA PECTORIS. See also: Bisoprolol Fumarate (has salt form). Drug Indication Bisoprolol is indicated for the treatment of mild to moderate hypertension. It may be used off-label to treat heart failure, atrial fibrillation, and angina pectoris. Mechanism of Action Though the mechanism of action of bisoprolol has not been fully elucidated in hypertension, it is thought that therapeutic effects are achieved through the antagonism of β-1adrenoceptors to result in lower cardiac output. Bisoprolol is a competitive, cardioselective β1-adrenergic antagonist. When β1-receptors (located mainly in the heart) are activated by adrenergic neurotransmitters such as epinephrine, both the blood pressure and heart rate increase, leading to greater cardiovascular work, increasing the demand for oxygen. Bisoprolol reduces cardiac workload by decreasing contractility and the need for oxygen through competitive inhibition of β1-adrenergic receptors. Bisoprolol is also thought to reduce the output of renin in the kidneys, which normally increases blood pressure. Additionally, some central nervous system effects of bisoprolol may include diminishing sympathetic nervous system output from the brain, decreasing blood pressure and heart rate. Therapeutic Uses Adrenergic beta-1 Receptor Antagonists; Antihypertensive Agents; Sympatholytics /CLINICAL TRIALS/ ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. The Web site is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each ClinicalTrials.gov record presents summary information about a study protocol and includes the following: Disease or condition; Intervention (for example, the medical product, behavior, or procedure being studied); Title, description, and design of the study; Requirements for participation (eligibility criteria); Locations where the study is being conducted; Contact information for the study locations; and Links to relevant information on other health Web sites, such as NLM's MedlinePlus for patient health information and PubMed for citations and abstracts for scholarly articles in the field of medicine. Bisoprolol is included in the database. Zebeta is indicated in the management of hypertension. It may be used alone or in combination with other antihypertensive agents. /Included in US product label/ MEDICATION (VET): Bisoprolol is a beta1 blocker that is somewhat cardioselective and therefore is indicated for conditions that require a reduction in heart rate, heart conductivity, or contractility. Such conditions include tachyarrhythmias and atrial fibrillation. In people it is used to treat hypertension, but this use has not been explored in animals. For more Therapeutic Uses (Complete) data for Bisoprolol (6 total), please visit the HSDB record page. Drug Warnings Zebeta is contraindicated in patients with cardiogenic shock, overt cardiac failure, second or third degree AV block, and marked sinus bradycardia. VET: Use cautiously in animals with airway disease, myocardial failure, and cardiac conduction disturbances. Use cautiously in animals with low cardiac reserve. Special caution should be exercised when administering bisoprolol fumarate to patients with a history of severe heart failure. Safety and effectiveness of bisoprolol doses higher than 10 mg/day in patients with heart failure have not been established. Sympathetic stimulation is a vital component supporting circulatory function in congestive heart failure and inhibition with beta-blockade always carries the potential hazard of further depressing myocardial contractility and precipitating cardiac failure. In general beta-blocking agents should be avoided in patients with overt congestive heart failure. However, in some patients with compensated cardiac failure, it may be necessary to utilize them. In such a situation, they must be used cautiously. Bisoprolol fumarate acts selectively without abolishing the effects of digitalis. However, the positive inotropic effect of digitalis may be reduced by the negative inotropic effect of bisoprolol fumarate when the two drugs are used concomitantly. The effects of beta-blockers and digitalis are additive in depressing A-V conduction. Exacerbation of angina pectoris, and, in some instances, myocardial infarction or ventricular arrhythmia, have been observed in patients with coronary artery disease following abrupt cessation of therapy with beta-blockers. Such patients should, therefore, be cautioned against interruption or discontinuation of therapy without the physician's advice. Even in patients without overt coronary artery disease, it may be advisable to taper therapy with Zebeta over approximately one week with the patient under careful observation. If withdrawal symptoms occur, Zebeta therapy should be reinstituted, at least temporarily. For more Drug Warnings (Complete) data for Bisoprolol (17 total), please visit the HSDB record page. Pharmacodynamics Bisoprolol decreases heart rate (chronotropy), decreases contractility (inotropy), and reduces blood pressure. The results of various clinical studies indicate that bisoprolol reduces cardiovascular mortality and all-cause mortality in patients with heart failure and decreased cardiac ejection fraction (EF). |
| 分子式 |
C18H31NO4
|
|---|---|
| 分子量 |
325.44
|
| 精确质量 |
325.225
|
| 元素分析 |
C, 66.43; H, 9.60; N, 4.30; O, 19.66
|
| CAS号 |
66722-44-9
|
| 相关CAS号 |
Bisoprolol-d5; 1189881-87-5; Bisoprolol hemifumarate; 104344-23-2; Bisoprolol fumarate; 105878-43-1
|
| PubChem CID |
2405
|
| 外观&性状 |
Colorless to light yellow liquid
|
| 密度 |
1.0±0.1 g/cm3
|
| 沸点 |
445.0±45.0 °C at 760 mmHg
|
| 熔点 |
100-103
100 °C |
| 闪点 |
222.9±28.7 °C
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
| 折射率 |
1.500
|
| LogP |
2.14
|
| tPSA |
59.95
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
12
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
278
|
| 定义原子立体中心数目 |
0
|
| SMILES |
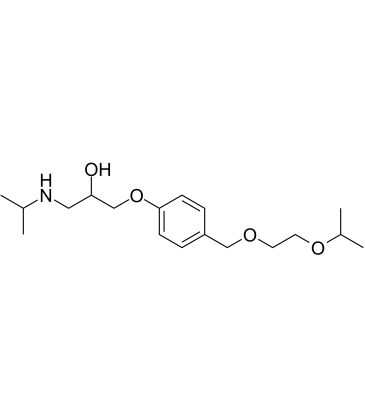
CC(OCCOCC1=CC=C(OCC(CNC(C)C)O)C=C1)C
|
| InChi Key |
VHYCDWMUTMEGQY-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C18H31NO4/c1-14(2)19-11-17(20)13-23-18-7-5-16(6-8-18)12-21-9-10-22-15(3)4/h5-8,14-15,17,19-20H,9-13H2,1-4H3
|
| 化学名 |
1-(propan-2-ylamino)-3-[4-(2-propan-2-yloxyethoxymethyl)phenoxy]propan-2-ol
|
| 别名 |
CL297,939; CL-297,939; Bisoprolol; CL 297,939
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0728 mL | 15.3638 mL | 30.7276 mL | |
| 5 mM | 0.6146 mL | 3.0728 mL | 6.1455 mL | |
| 10 mM | 0.3073 mL | 1.5364 mL | 3.0728 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03278509 | Active Recruiting |
Drug: Metoprolol Succinate Drug: Bisoprolol |
Acute Myocardial Infarction ST Elevation Myocardial Infarction |
Karolinska Institutet | September 11, 2017 | Phase 4 |
| NCT03917914 | Active Recruiting |
Drug: Bisoprolol Drug: Placebo Oral Tablet |
Cardiovascular Diseases Chronic Obstructive Pulmonary Disease |
The George Institute | June 30, 2020 | Phase 3 |
| NCT05794997 | Active Recruiting |
Drug: Propranolol or Carvedilol Drug: Atenolol, Bisoprolol or Sotalol |
Hypertension | Brigham and Women's Hospital | November 30, 2022 | N/A |
| NCT05540600 | Recruiting | Drug: Digoxin 0.25 mg Drug: Bisoprolol |
Atrial Fibrillation Left Atrial Rhythm |
University of Monastir | September 12, 2022 | Phase 3 |
| NCT05294887 | Recruiting | Drug: Bisoprolol Drug: Diltiazem Drug: Placebo |
Microvascular Angina Vasospastic Angina Prinzmetal Angina |
Charite University, Berlin, Germany |
March 4, 2022 | Phase 4 |
|
|
 Mibenratide TFA
Mibenratide TFA
 Clorprenaline-d7
Clorprenaline-d7
 β2ARagonist 2
β2ARagonist 2
 (R)-9-Hydroxy Risperidone-d4
(R)-9-Hydroxy Risperidone-d4
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA


