| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
HIV-1
|
|---|---|
| 体外研究 (In Vitro) |
bictegravir (BIC) 抑制链转移活性,IC50 为 7.5± 0.3 nM。 bictegravir 的 IC50 为 241±51 nM,对 HIV-1 IN 3'-加工活性的抑制作用比对链转移活性的抑制作用弱得多。与模拟处理的对照相比,bictegravir 使 2-LTR 环的积累增加了大约五倍,并将感染细胞中真实整合产物的数量减少了 100 倍。bictegravir 的 EC50 分别为 1.5 和 2.4 nM,有效抑制 MT-2 和 MT-4 细胞中的 HIV-1 复制。 bictegravir 的 EC50 分别为 1.5±0.3 nM 和 6.6±4.1 nM,在原代 CD4+ T 淋巴细胞和单核细胞衍生的巨噬细胞中表现出强大的抗病毒作用。这些值与 T 细胞系中观察到的值一致。
|
| 体内研究 (In Vivo) |
HIV-1 IIIb 在 MT-2 细胞上于 37°C 下批量培养 3 小时,细胞密度为 2×106 个细胞/mL。将 Bictegravir (BIC) 或 DMSO(模拟处理对照)给予感染的 MT-2 细胞,其最终浓度至少是每种药物抗病毒 50% 有效浓度 (EC50) 的 20 倍。收集细胞以获得总 DNA将这些板在 37°C 下孵育 12 小时(用于后期逆转录产物定量)或 24 小时(用于 2-LTR 环和 Alu-LTR 产物定量)后进行分离。使用 DNA 微型试剂盒,从每个孔中提取 DNA 并收集为 100 μL 洗脱液。每个样本中的宿主珠蛋白基因水平用于标准化 TaqMan 实时 PCR 定量的 2-LTR 连接点(2-LTR 环)、晚期逆转录产物和整合连接点 (Alu-LTR)[1]。
|
| 细胞实验 |
在批量培养中,MT-2 细胞在 37°C 下用 HIV-1 IIIb 感染 3 小时,细胞密度为 2×106 个细胞/mL。将 Bictegravir (BIC) 或 DMSO(模拟处理对照)给予感染的 MT-2 细胞,其最终浓度至少是每种药物抗病毒 50% 有效浓度 (EC50) 的 20 倍。将这些板在 37°C 下孵育 12 小时(用于后期逆转录产物定量)或 24 小时(用于 2-LTR 环和 Alu-LTR 产物定量)后,收获细胞以进行总 DNA 分离。使用 DNA 微型试剂盒,从每个孔中提取 DNA 并收集为 100 μL 洗脱液。每个样本中的宿主球蛋白基因水平作为 TaqMan 实时 PCR 定量 2-LTR 连接(2-LTR 环)、晚期逆转录产物和整合连接(Alu-LTR)的标准[1]。
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Bictegravir is rapidly absorbed within the body. Tmax= 2.0-4.0h BIC is mainly eliminated through UGT1A1 glucuronidation and CYP3A4 oxidation, equally. About 1% of the bictegravir dose is excreted in the urine, unchanged. 0.2 L/Kg in humans Bictegravir is mainly cleared by the kidneys. Those with renal clearance <30 should not take bictegravir [FDA LABEL] Metabolism / Metabolites In a 10-day dose-ranging study, monotherapy (5 mg to 100 mg) once daily in adults who were not previously treated with bictegravir, the median half-life of BIC ranged from 15.9 h - 20.9 h. Bictegravir is metabolized in the liver and kidneys. CYP3A4 and UGT1A are the primary enzymes involved in the metabolism of bictegravir. Administration of bictegravir is not advised in patients with renal creatinine clearance of <30 mL/min and patients with hepatic disease [FDA LABEL]. Biological Half-Life Half-life is 17.3 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In large clinical trials, therapy with bictegravir combined with emtricitabine and tenofovir alafenamide was associated with alanine aminotransferase (ALT) elevations (above 1.5 times ULN) in 11% patients, but these rates were similar to those in comparator groups (12% to 15%) receiving matched background optimized antiretroviral therapy without bictegravir. Elevations above 5 times ULN occurred in only 1.4% of bictegravir vs 0.9% to 1.3% of control comparator arm subjects. The elevations were not associated with clinical symptoms and generally did not require dose modification. In addition, there were no instances of acute hepatocellular liver injury with jaundice. The product label for bictegravir mentions acute exacerbations of hepatitis B and hepatic failure as potential adverse reactions when bictegravir with emtricitabine and tenofovir is discontinued. This adverse reaction can occur upon discontinuation of any antiretroviral regimen with concurrent activity against HBV and represents the effects of tenofovir and emtricitabine. Nevertheless, since its approval and its more widescale use, there have been no published reports of clinically apparent cases of liver injury or exacerbation of hepatitis B convincingly attributed to bictegravir. Likelihood score: E* (unproven but suspected potential cause of liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Limited information indicates that maternal bictegravir 50 mg once daily produce low levels in milk and infant serum. Until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. Achieving and maintaining viral suppression with antiretroviral therapy decreases breastfeeding transmission risk to less than 1%, but not zero. Individuals with HIV who are on antiretroviral therapy with a sustained undetectable viral load and who choose to breastfeed should be supported in this decision. If a viral load is not suppressed, banked pasteurized donor milk or formula is recommended. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Gynecomastia has been reported among men receiving highly active antiretroviral therapy. Gynecomastia is unilateral initially, but progresses to bilateral in about half of cases. No alterations in serum prolactin were noted and spontaneous resolution usually occurred within one year, even with continuation of the regimen. Some case reports and in vitro studies have suggested that protease inhibitors might cause hyperprolactinemia and galactorrhea in some male patients, although this has been disputed. The relevance of these findings to nursing mothers is not known. The prolactin level in a mother with established lactation may not affect her ability to breastfeed. Protein Binding > 99 % bound to human plasma Blood to plasma ratio: 0.64 |
| 参考文献 |
[1]. Antimicrob Agents Chemother.2016 Nov 21;60(12):7086-7097.
|
| 其他信息 |
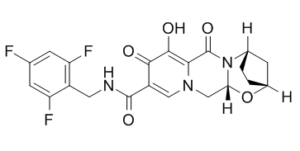
Description
Bictegravir is a monocarboxylic acid amide obtained by formal condensation of the carboxy group of (2R,5S,13aR)-8-hydroxy-7,9-dioxo-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxylic acid with the amino group of 2,4,6-trifluorobenzylamine. It is a second-generation integrase strand transfer inhibitor (INSTI) and used (as its sodium salt) for the treatment of HIV-1. It has a role as a HIV-1 integrase inhibitor. It is a monocarboxylic acid amide, a secondary carboxamide, a trifluorobenzene and an organic heterotetracyclic compound. It is a conjugate acid of a bictegravir(1-). Bictegravir is a recently approved investigational drug that has been used in trials studying the treatment of HIV-1 and HIV-2 infection. It has been approved for HIV-1 monotherapy combined with 2 other antiretrovirals in a single tablet. Bictegravir is a human immunodeficiency virus (HIV) integrase strand transfer inhibitor, the fourth in this class of agents that target the viral integrase. Bictegravir is used only in combination with other antiretroviral agents in the treatment of HIV infection and it has had limited use. Bictegravir is associated with a low rate of serum aminotransferase elevations during therapy, but has not been linked to instances of acute, clinically apparent liver injury. Bictegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI), that is used to treat HIV infection. Upon oral administration, bictegravir inhibits the strand transfer activity of HIV-1 integrase, an HIV-1 coded enzyme that is necessary for viral replication. Inhibition of integrase prevents the integration of linear HIV-1 DNA into host genomic DNA. Drug Indication Bictegravir is indicated in the management of HIV-1 infection in patients not previously treated with antiretroviral therapy. Additionally, Bictegravir is indicated in the management of HIV-1 infection in patients who are virologically suppressed (HIV-1 RNA <50 c/mL) on a regular antiretroviral regimen for a minimum of three months without a history of failure in treatment and no known factors associated with the resistance to the individual components of the medication. It is used in combination with tenofovir and emtricitabine. FDA Label Mechanism of Action This single dose medication inhibits the strand transfer of viral DNA into the human genome, preventing HIV-1 virus replication and propagation. In vitro, bictegravir has shown powerful antiviral activity against HIV-2 and various subtypes of HIV-1. It has shown synergistic effects when combined with other ARVs, including tenofovir alafenamide (TAF), emtricitabine (FTC), and darunavir (DRV). The three components of the first USA approved medication ( trade name: Biktarvy ) are as follows: Bictegravir: integrase strand transfer inhibitor; INSTI), an HIV-1 encoded enzyme necessary for viral replication. Inhibition of the integrase enzyme prevents the integration of HIV-1 into host DNA, blocking the conversion of the HIV-1 provirus and progression of the virus [FDA LABEL]. Emtricitabine: FTC, is phosphorylated by cellular enzymes to form emtricitabine 5'-triphosphate. Emtricitabine is phosphorylated to form emtricitabine 5'-triphosphate intracellularly. This metabolite inhibits the activity of human immunodeficiency virus (HIV) reverse transcriptase by competing with the substrate deoxycytidine 5'-triphosphate and by incorporating itself into viral DNA preventing DNA chain elongation [FDA LABEL]. Tenofovir Alafenamide: TAF is a phosphonamidate prodrug of tenofovir (2′-deoxyadenosine monophosphate analog). Plasma exposure to TAF leads to leakage into cells and then TAF is intracellularly converted to tenofovir by hydrolysis by cathepsin. Tenofovir is subsequently phosphorylated by cellular kinases to the metabolite tenofovir diphosphate, which is the active form of the drug. Tenofovir diphosphate inhibits HIV-1 replication by incorporating into viral DNA by the HIV reverse transcriptase, resulting in DNA chain-termination. Tenofovir diphosphate also weakly inhibits mammalian DNA polymerases [FDA LABEL]. |
| 分子式 |
C21H18F3N3O5
|
|
|---|---|---|
| 分子量 |
449.37
|
|
| 精确质量 |
449.119
|
|
| 元素分析 |
C, 56.13; H, 4.04; F, 12.68; N, 9.35; O, 17.80
|
|
| CAS号 |
1611493-60-7
|
|
| 相关CAS号 |
Bictegravir sodium;1807988-02-8;Bictegravir-15N,d2
|
|
| PubChem CID |
90311989
|
|
| 外观&性状 |
Solid powder
|
|
| 密度 |
1.62±0.1 g/cm3
|
|
| 沸点 |
682.5±55.0 °C at 760 mmHg
|
|
| 闪点 |
366.6±31.5 °C
|
|
| 蒸汽压 |
0.0±2.2 mmHg at 25°C
|
|
| 折射率 |
1.664
|
|
| LogP |
-1.26
|
|
| tPSA |
99.2
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
9
|
|
| 可旋转键数目(RBC) |
3
|
|
| 重原子数目 |
32
|
|
| 分子复杂度/Complexity |
912
|
|
| 定义原子立体中心数目 |
3
|
|
| SMILES |
FC1C=C(C=C(C=1CNC(C1C(C(=C2C(N3[C@@H](CN2C=1)O[C@@H]1CC[C@H]3C1)=O)O)=O)=O)F)F
|
|
| InChi Key |
SOLUWJRYJLAZCX-LYOVBCGYSA-N
|
|
| InChi Code |
InChI=1S/C21H18F3N3O5/c22-9-3-14(23)12(15(24)4-9)6-25-20(30)13-7-26-8-16-27(10-1-2-11(5-10)32-16)21(31)17(26)19(29)18(13)28/h3-4,7,10-11,16,29H,1-2,5-6,8H2,(H,25,30)/t10-,11+,16+/m0/s1
|
|
| 化学名 |
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : 83.3 ~90 mg/mL ( 185.37 ~200.27 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.56 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.56 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.56 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.5 mg/mL (5.56 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 5 中的溶解度: 10% DMSO+40% PEG300+5% Tween-80+45% Saline: ≥ 2.5 mg/mL (5.56 mM) 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2253 mL | 11.1267 mL | 22.2534 mL | |
| 5 mM | 0.4451 mL | 2.2253 mL | 4.4507 mL | |
| 10 mM | 0.2225 mL | 1.1127 mL | 2.2253 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Resistance profile of BIC and other INSTIs against 47 HIV-1 patient-derived isolates with INSTI resistance mutations.Antimicrob Agents Chemother. 2016 Dec; 60(12): 7086–7097. |
|---|
Progress of BIC, DTG, and EVG resistance selection with HIV-1 IIIb.Antimicrob Agents Chemother. 2016 Dec; 60(12): 7086–7097. |
HIV-1 IIIb resistance breakthrough in MT-2 cells. Viral resistance breakthrough for each drug was tested in four independent infected cultures in the presence of constant drug pressure for up to 35 days.Antimicrob Agents Chemother. 2016 Dec; 60(12): 7086–7097. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

























 COA
COA