| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| Other Sizes |
|

| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
The steady-state of berotralstat is reached within 6 to 12 days following initial administration. After once-daily administration, the Cmax and AUC of berotralstat at steady-state is approximately five times that of the drug after a single dose. Following oral administration of berotralstat once-daily, the steady-state Cmax was 158 ng/mL (range: 110 to 234 ng/mL) at the dose of 150 mg and 97.8 ng/mL (range: 63 to 235 ng/mL) at the dose of 110 mg. The area under the curve over the dosing interval (AUCtau) was 2770 ng*hr/mL (range: 1880 to 3790 ng*hr/mL) and 1600 ng*hr/mL (range: 950 to 4170 ng*hr/mL) at the dose of 110 mg. The median Tmax is 2 hours in a fasted state and a high-fat meal delays the Tmax to 5 hours. The Tmax can range from 1 to 8 hours. Following a single oral dose administration of 300 mg radiolabeled berotralstat, approximately 9% of the drug was excreted in the urine, where 1.8 to 4.7% of the total radiolabeled compound accounted for the unchanged parent drug. About 79% of the drug was excreted in feces. The blood to plasma ratio was approximately 0.92 following a single 300 mg dose administration of radiolabeled berotralstat. There is no information on the clearance rate. Metabolism / Metabolites Berotralstat is metabolized by CYP2D6 and CYP3A4. The metabolic pathway and the metabolites of berotralstat have not yet been characterized. Following a single oral dose administration of 300 mg radiolabeled berotralstat, about 34% of the total plasma radioactivity accounted for the unchanged drug while about eight detectable metabolites accounted for 1.8 to 7.8% of the total radioactivity. Biological Half-Life Following a single oral dose administration of 300 mg radiolabeled berotralstat, the median elimination half-life of berotralstat was approximately 93 hours, ranging from 39 to 152 hours. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In preregistration trials, mild, transient serum aminotransferase elevations occurred in 2% to 5% of patients receiving berotralstat vs 10% of those on placebo. Values above 5 times the upper limit of normal (ULN) were uncommon (less than 1%). Furthermore, bilirubin levels remained normal and no patient developed symptomatic acute liver injury. In many instances, other causes for the liver test abnormalities were present including preexisting nonalcoholic fatty liver disease, chronic viral hepatitis, or gallstone disease. Nevertheless, occasional patients required drug discontinuation because of liver test abnormalities. Since its licensure and more widescale clinical use, there have been no published reports of acute liver injury attributed to berotralstat. Likelihood score: E* (unproven but suspected rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Berotralstat is a plasma kallikrein inhibitor indicated for prophylaxis to prevent attacks of hereditary angioedema. No information is available on the excretion of berotralstat into breastmilk. Because berotralstat is about 99% bound to plasma proteins, the amounts in milk are likely to be very low. If berotralstat is required by the mother, it is not a reason to discontinue breastfeeding. Until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Plasma protein binding is approximately 99%. |
| 参考文献 |
: Sohtome Y, Sodeoka M. Development of Chaetocin and S-Adenosylmethionine Analogues as Tools for Studying Protein Methylation. Chem Rec. 2018 Dec;18(12):1660-1671.
|
| 其他信息 |
Berotralstat is a selective inhibitor of plasma kallikrein used in the prophylaxis of attacks of hereditary angioedema (HAE). It works by blocking the enzymatic activity of plasma kallikrein in releasing bradykinin, the major biologic peptide that promotes swelling and pain associated with attacks of HAE. Berotralstat is strictly used to prevent, but not treat, these attacks. Developed by BioCryst Pharmaceuticals, berotralstat is marketed under the name Orladeyo as oral capsules. Berotralstat was first approved by the FDA on December 3, 2020, as the first once-daily oral therapy to prevent angioedema attacks of HAE in adults and pediatric patients 12 years and older. Berotralstat was approved by the European Commission on April 30, 2021 and by Health Canada on June 06, 2022.
Berotralstat is a Plasma Kallikrein Inhibitor. The mechanism of action of berotralstat is as a Kallikrein Inhibitor, and Cytochrome P450 2D6 Inhibitor, and Cytochrome P450 3A4 Inhibitor, and P-Glycoprotein Inhibitor. Berotralstat is a small molecule inhibitor of plasma kallikrein that is used to prevent acute attacks of hereditary angioedema (HAE) in adults and children 12 years of age or older. Berotralstat has been linked to occasional mild-to-moderate elevations in serum aminotransferase levels during therapy but has not been implicated in instances of clinically apparent liver injury with symptoms or jaundice. See also: Berotralstat Hydrochloride (active moiety of). Drug Indication Berotralstat is indicated for prophylaxis of attacks of hereditary angioedema (HAE) in adults and pediatric patients 12 years and older. It is not used for the treatment of acute HAE attacks. Orladeyo is indicated for routine prevention of recurrent attacks of hereditary angioedema (HAE) in adult and adolescent patients aged 12 years and older. Treatment of hereditary angioedema Mechanism of Action Hereditary angioedema (HAE) is a rare genetic disorder associated with severe swelling of the skin and upper airway. It is caused by mutations in the regulatory or coding regions of the gene that encodes C1 inhibitor (SERPING1), which result in either a deficiency (type I) or dysfunction (type II) of C1 inhibitor (C1 esterase inhibitor, C1-INH). C1 inhibitor is a serine protease inhibitor that normally regulates bradykinin production by covalently binding to and inactivating plasma kallikrein. Plasma kallikrein is a protease that cleaves high-molecular-weight-kininogen (HMWK) to generate cleaved HMWK (cHMWK). During HAE attacks, the levels of plasma kallikrein fall, leading to the cleavage of high-molecular-weight-kininogen and the release of bradykinin, a potent vasodilator that increases vascular permeability. Bradykinin plays a major role in promoting edema and pain associated with HAE. Patients with HAE cannot properly regulate plasma kallikrein activity due to the deficiency or dysfunction of a serum inhibitor of C1 inhibitor, leading to uncontrolled increases in plasma kallikrein activity and recurrent angioedema attacks. Berotralstat is a potent inhibitor of plasma kallikrein that works by binding to plasma kallikrein and blocking its proteolytic activity, thereby controlling excess bradykinin generation. Pharmacodynamics Berotralstat prevents angioedema attacks by inhibiting plasma kallikrein, thereby regulating excess bradykinin generation in patients with hereditary angioedema (HAE). It had a fast onset of action, long duration of action, and acceptable tolerance in clinical trials. Berotralstat inhibits plasma kallikrein in a concentration-dependent. In clinical trials, berotralstat reduced HAE attack rates at 24 weeks, and its effects sustained through 48 weeks. In clinical trials, doses of berotralstat higher than 150 mg once daily led to QT Prolongation in a concentration-dependent manner. |
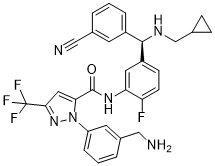
| 分子式 |
C30H26F4N6O
|
|---|---|
| 分子量 |
562.560659885406
|
| 精确质量 |
562.21
|
| CAS号 |
1809010-50-1
|
| 相关CAS号 |
Berotralstat dihydrochloride;1809010-52-3
|
| PubChem CID |
137528262
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
4.5
|
| tPSA |
109
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
41
|
| 分子复杂度/Complexity |
938
|
| 定义原子立体中心数目 |
1
|
| SMILES |
FC1C=CC(=CC=1NC(C1=CC(C(F)(F)F)=NN1C1C=CC=C(CN)C=1)=O)[C@@H](C1C=CC=C(C#N)C=1)NCC1CC1
|
| InChi Key |
UXNXMBYCBRBRFD-MUUNZHRXSA-N
|
| InChi Code |
InChI=1S/C30H26F4N6O/c31-24-10-9-22(28(37-17-18-7-8-18)21-5-1-3-19(11-21)15-35)13-25(24)38-29(41)26-14-27(30(32,33)34)39-40(26)23-6-2-4-20(12-23)16-36/h1-6,9-14,18,28,37H,7-8,16-17,36H2,(H,38,41)/t28-/m1/s1
|
| 化学名 |
1-[3-(aminomethyl)phenyl]-N-(5-{(R)-(3-
cyanophenyl)[(cyclopropylmethyl)amino]methyl}-2-
fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-
carboxamide
|
| 别名 |
ORLADEYO BCX-7353 BCX 7353BCX7353
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~177.76 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 8.5 mg/mL (15.11 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 85.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7776 mL | 8.8879 mL | 17.7759 mL | |
| 5 mM | 0.3555 mL | 1.7776 mL | 3.5552 mL | |
| 10 mM | 0.1778 mL | 0.8888 mL | 1.7776 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1




























