| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
In a study, three patients were administered 50 mg of radiolabelled 14C-benserazide by both intravenous and oral routes. Three additional patients received oral doses of 50 mg 14C-benserazide alone. Comparison of the time-plasma concentration curves of total radioactivity in the patients receiving oral and intravenous 14C-benserazide indicated that between 66% and 74% of the administered dose was absorbed from the gastrointestinal tract. Peak plasma concentrations of radioactivity were detected one hour after oral administration in five of the six patients. Benserazide is rapidly excreted in the urine in the form of metabolites, mostly within the first 6 hours of administration, 85% of urinary excretion occurs within 12 hours. Elimination of radiolabelled 14C-benserazide was primarily by urinary excretion with 86% to 90% of an intravenous dose recovered in the urine while 53% to 64% of an oral dose was detected in the urine. The majority of the 14C-benserazide was ultimately accounted for in the urine within 48 hours after administration. Fecal recovery studies conducted over five to eight days accounted for the majority (about 30%) of the remainder of the administered 14C-benserazide. Ultimately, benserazide is almost entirely eliminated by metabolism. These metabolites are mainly excreted in the urine (64%) and to a smaller extent in the feces (24%). Readily accessible data regarding the volume of distribution of benserazide is not available. Readily accessible data regarding the clearance of benserazide is not available. Metabolism / Metabolites Benserazide is hydroxylated to trihydroxybenzylhydrazine in the intestinal mucosa and the liver. Trihydroxybenzylhydrazine is a potent inhibitor of the aromatic acid decarboxylase, and it is believed that the levodopa in a levodopa/benserazide combination product is largely protected against decarboxylation mainly by way of this benserazide metabolite. Biological Half-Life The half-life of benserazide is documented as 1.5 hours. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Protein Binding
Benserazide is observed as experiencing 0% protein binding. |
| 参考文献 | |
| 其他信息 |
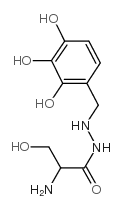
Benserazide is a carbohydrazide that results from the formal condensation of the carboxy group of DL-serine with the primary amino group of 4-(hydrazinylmethyl)benzene-1,2,3-triol. An aromatic-L-amino-acid decarboxylase inhibitor (DOPA decarboxylase inhibitor) that does not enter the central nervous system, it is used as its hydrochloride salt as an adjunct to levodopa in the treatment of parkinsonism. By preventing the conversion of levodopa to dopamine in the periphery, it causes an increase in the amount of levodopa reaching the central nervous system and so reduces the required dose. Benserazide has no antiparkinson actions when given alone. It has a role as an EC 4.1.1.28 (aromatic-L-amino-acid decarboxylase) inhibitor, an antiparkinson drug and a dopaminergic agent. It is a carbohydrazide, a member of catechols, a primary amino compound and a primary alcohol. It is a conjugate base of a benserazide(1+).
When levodopa is used by itself as a therapy for treating Parkinson's disease, its ubiquitous metabolism into dopamine is responsible for a resultant increase in the levels of circulating dopamine in the blood and to various extracerebral tissues. This can result in a number of side effects like nausea, vomiting, or even cardiac arrhythmias that may diminish patient adherence. A decarboxylase inhibitor like benserazide is consequently an effective compound to combine with levadopa as it is incapable of crossing the blood-brain barrier itself but acts to prevent the formation of dopamine from levadopa in extracerebral tissues - thereby acting to minimize the occurrence of extracerebral side effects. Levodopa/benserazide combination products are used commonly worldwide for the management of Parkinson's disease. In particular, although the specific levodopa/benserazide combination is formally approved for use in Canada and much of Europe, the FDA has approved another similar levodopa/dopa decarboxylase inhibitor combination in the form of levodopa and carbidopa. Moreover, the European Medcines Agency has conferred an orphan designation upon benseraside since 2015 for its potential to be used as a therapy for beta thalassaemia as well. An inhibitor of DOPA DECARBOXYLASE that does not enter the central nervous system. It is often given with LEVODOPA in the treatment of parkinsonism to prevent the conversion of levodopa to dopamine in the periphery, thereby increasing the amount that reaches the central nervous system and reducing the required dose. It has no antiparkinson actions when given alone. Drug Indication The primary therapeutic use for which benserazide is currently indicated for is as a combination therapy with levadopa for the treatment of Parkinson's disease in adults > 25 years of age, with the exception of drug-induced parkinsonism. At certain doses, the combination product of levodopa and benserazide may also be used to treat restless legs syndrome, which is sometimes associated with Parkinson's disease. There have also been some studies that have prompted the European Medicines Agency to confer orphan designation upon benserazide hydrochloride as a potential therapy for beta thalassaemia. Although studies are ongoing, no evidence has been formally elucidated as of yet. Mechanism of Action The combination of levodopa and benserazide is an anti-Parkinsonian agent. Levodopa itself is the metabolic precursor of dopamine. In Parkinson's disease, dopamine is depleted to a large degree in the striatum, pallidum, and substantia nigra in the central nervous system (CNS). The administration of levodopa to treat the disease is subsequently proposed to facilitate raises in the levels of available dopamine in these areas. The metabolism of levodopa to dopamine occurs via the enzyme dopa decarboxylase, although unfortunately, this metabolism can also occur in extracerebral tissues. As a result, the full therapeutic effect of an administered dose of levodopa may not be obtained if portions of it are catabolized outside of the CNS and various patient adherence diminishing extracerebral side effects due to the extracerebral presence of dopamine like nausea, vomiting, or even cardiac arrhythmias can also happen. Subsequently, a peripheral decarboxylase inhibitor like benserazide, which blocks the extracerebral decarboxylation of levodopa, when administered in combination with levodopa has obvious and significant advantages. Such benefits include reduced gastrointestinal side effects, a more rapid and complete response at the initiation of therapy, and a simpler dosing regimen. It is important to note, however, that benserazide is hydroxylated to trihydroxybenzylhydrazine in the intestinal mucosa and the liver, and that as a potent inhibitor of the aromatic amino acid decarboxylase, it is this trihydroxybenzylhydrazine metabolite of benserazide that mainly protects levodopa against decarboxylation to dopamine in the gut and also around the rest of the body outside of the blood-brain barrier. Regardless, because Parkinson's disease progresses even with the therapy of levodopa and benserazide, this kind of combined therapy is only ever indicated if it is capable of improving the quality of life and adverse effect profile of using such drugs for Parkinson's patients and there is little to be gained by switching to or starting this combination therapy if patients are already being managed with stable, effective, and well-tolerated levadopa-only therapy. Finally, it is also proposed that benserazide hydrochloride may be able to treat beta thalassaemia by maintaining the active expression of the gene for fetal hemoglobin so that constant production of fetal hemoglobin may replace the missing adult hemoglobin variation that is characteristic of patients with the condition, thereby decreasing the need for blood transfusion therapy. |
| 分子式 |
C10H15N3O5
|
|---|---|
| 分子量 |
257.2432
|
| 精确质量 |
257.101
|
| CAS号 |
322-35-0
|
| 相关CAS号 |
Benserazide hydrochloride;14919-77-8
|
| PubChem CID |
2327
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.541 g/cm3
|
| 沸点 |
574.2ºC at 760 mmHg
|
| 闪点 |
301ºC
|
| 折射率 |
1.678
|
| LogP |
-1.3
|
| tPSA |
148.07
|
| 氢键供体(HBD)数目 |
7
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
278
|
| 定义原子立体中心数目 |
0
|
| SMILES |
[2H]C(C([2H])(O)[2H])(N)C(NNCC1=C(O)C(O)=C(O)C=C1)=O.Cl
|
| InChi Key |
BNQDCRGUHNALGH-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C10H15N3O5/c11-6(4-14)10(18)13-12-3-5-1-2-7(15)9(17)8(5)16/h1-2,6,12,14-17H,3-4,11H2,(H,13,18)
|
| 化学名 |
2-amino-3-hydroxy-N'-[(2,3,4-trihydroxyphenyl)methyl]propanehydrazide
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.8874 mL | 19.4371 mL | 38.8742 mL | |
| 5 mM | 0.7775 mL | 3.8874 mL | 7.7748 mL | |
| 10 mM | 0.3887 mL | 1.9437 mL | 3.8874 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























