| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|

| 靶点 |
DNMT1; pyrimidine nucleoside analogue of cytidine; Nucleoside Antimetabolite; Autophagy
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
与许多基因相关的未甲基化 CpG 岛在恶性肿瘤中部分或完全甲基化,并且可以被 5-氮杂胞苷重新激活 [1]。 5-Azacytidine 在影响 DNA 甲基转移酶活性的相同剂量范围内作为 Friend 红白血病细胞红系分化的温和诱导剂 [2]。 5-Azacytidine 抑制 L1210 细胞,ID50 和 ID90 值分别为 0.019 和 ~0.15 μg/mL [3]。
|
||
| 体内研究 (In Vivo) |
当大鼠接受 5-氮杂胞苷(100 mg/kg,腹腔注射)治疗两个小时或更长时间时,TdR-3H 掺入显着减少 [3]。
我们在癌症免疫活性小鼠模型中测试了这一假设,发现在体内,5-氮杂胞苷(AZA)和α-二氟甲基鸟氨酸(DFMO),无论是单独使用还是联合使用,都显著提高了存活率,降低了肿瘤负担,并导致激活的(IFNγ+)CD4+T细胞、CD8+T细胞和NK细胞的募集。与单药治疗相比,联合治疗的存活率显著提高,尽管招募的淋巴细胞差异较小。相反,联合治疗导致免疫抑制细胞如M2极化巨噬细胞显著减少,杀死肿瘤的M1巨噬细胞增加。在该模型中,用CSF1R阻断抗体耗竭巨噬细胞降低了AZA+DFMO治疗的疗效,并导致肿瘤微环境中M1巨噬细胞减少。这些观察结果表明,我们的新型联合疗法改变了肿瘤微环境中的巨噬细胞极化,招募了M1巨噬细胞并延长了存活时间[5]。 |
||
| 酶活实验 |
用抗白血病药物5-氮杂胞苷和5-氮杂-2'-脱氧胞苷治疗Friend红白血病细胞会导致DNA甲基转移酶活性的快速、时间依赖性和剂量依赖性降低,以及明显甲基化不足的DNA的合成。由于这种DNA在体内至少部分甲基化,并且是体外甲基化的极好底物,因此类似物处理细胞中DNA的低甲基化似乎是由于DNA甲基转移酶的缺失,而不是由于5-氮杂胞嘧啶取代的DNA天生不能作为甲基受体。抑制DNA合成可以阻断DNA甲基转移酶活性的丧失,而RNA合成抑制剂则不能,这表明类似物必须掺入DNA中以介导其对酶的作用,并且DNA中5-氮杂胞嘧啶对胞嘧啶的轻微替代(约0.3%)足以灭活细胞中95%以上的酶。有几条证据表明,DNA修饰模式的变化与分化有关。在这方面,重要的是5-氮杂胞苷和5-氮杂-2'-脱氧胞苷在影响DNA甲基转移酶活性的相同浓度范围内,作为Friend红白血病细胞红系分化的弱诱导剂。为了进行分化,必须清除细胞中的药物。不到24小时后,DNA甲基转移酶活性恢复正常水平,48小时内,从细胞中分离的DNA未检测到甲基化不足。这可能部分解释了为什么5-氮杂胞苷和5-氮杂-2'-脱氧胞苷在不到15%的人群中诱导分化,尽管它们最初对DNA甲基化有深远影响[2]。
|
||
| 细胞实验 |
近年来,代谢综合征(MS)在人类和动物代谢医学中引起了人们的关注。胰岛素抵抗、炎症、高瘦素血症和高胰岛素血症对其定义至关重要。MS是一组复杂的代谢危险因素,它们共同对体内的多个器官、组织和细胞产生广泛的影响。脂肪干细胞(ASCs)是存在于MS期间发炎的脂肪组织内的多能干细胞群。研究表明,这些细胞在MS期间失去了其干性和多能干细胞性,这大大降低了它们的治疗潜力。它们遭受氧化应激、细胞凋亡和线粒体退化。因此,本研究的目的是在体外使这些细胞恢复活力,以提高其软骨分化的有效性。ASCs的药物治疗基于白藜芦醇和5-氮杂胞苷的预处理。我们使用免疫荧光、透射和扫描电镜、实时PCR和流式细胞术评估了这些物质是否能够逆转代谢综合征衍生的ASCs的衰老表型,并在早期改善其软骨分化。获得的结果表明,5-氮杂胞苷和白藜芦醇调节线粒体动力学、自噬和ER应激,导致代谢受损的ASC软骨生成增强。因此,在临床应用这些细胞之前,用5-氮杂胞苷和白藜芦醇对这些细胞进行预处理可能成为必要的干预措施,以增强其多能性和治疗潜力[4]。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Azacitidine is rapidly absorbed after subcutaneous administration. In adult patients with myelodysplastic syndrome given a single subcutaneous dose of 75 mg/m2 of azacitidine, the Cmax and Tmax were 750 ng/ml and 0.5 hours, respectively. Based on the area under the curve, the bioavailability of subcutaneous azacitidine relative to intravenous azacitidine is approximately 89%. In 21 patients with cancer given subcutaneous azacitidine, the AUC and Cmax were approximately dose-proportional between 25 and 100 mg/m2. Multiple subcutaneous or intravenous doses of azacitidine are not expected to result in drug accumulation. Azacitidine and its metabolites are mainly excreted through urine. In five cancer patients given radioactive azacitidine intravenously, the cumulative urinary excretion was 85% of the radioactive dose. Fecal excretion accounted for less than 1% of administered radioactivity over three days. Following the subcutaneous administration of 14C-azacitidine, the mean excretion of radioactivity in urine was 50%. In patients given an intravenous dose of azacitidine, the volume of distribution is 76 L. Azacitidine has an apparent subcutaneous clearance of 167 L/hour in adults. In pediatric patients, the geometric mean clearance was 21.8 L/hour. Metabolism / Metabolites An in vitro study of azacitidine incubation in human liver fractions indicated that cytochrome P450 (CYP) enzymes do not participate in the metabolism of azacitidine. Azacitidine is metabolized through spontaneous hydrolysis and deamination mediated by cytidine deaminase. An in vitro study of azacitidine incubation in human liver fractions indicated that azacitidine may be metabolized by the liver. The potential of azacitidine to inhibit cytochrome P450 (CYP) enzymes is not known. Route of Elimination: Following IV administration of radioactive azacitidine to 5 cancer patients, the cumulative urinary excretion was 85% of the radioactive dose. Fecal excretion accounted for <1% of administered radioactivity over three days. Mean excretion of radioactivity in urine following SC administration of 14C-azacitidine was 50%. Half Life: Mean elimination half-life is approximately 4 hours. Biological Half-Life The mean half-life of azacitidine after subcutaneous administration is 41 minutes. The mean elimination half-life of azacitidine and its metabolites was about 4 hours for intravenous and subcutaneous administrations. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In clinical trials, serum enzyme elevations occurred in up to 16% of patients on azacitidine therapy for cancer or myelodysplasia who had concurrent, underlying liver disease or liver metastases, but rarely in persons without a preexisting hepatic illness. In subsequent studies, liver adverse reactions attributed to azacitidine have rarely been reported, at least when it is given in conventional doses. Nevertheless, monitoring of serum enzyme levels is recommended in treating patients who have concurrent liver disease. Cases of clinically apparent liver injury attributed to azacitidine in patients without underlying liver disease have not been reported in the literature. Likelihood score: E* (unproven but suspected cause of clinically apparent liver injury, particularly in persons with pre-existing liver disease). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Most sources consider breastfeeding to be contraindicated during maternal antineoplastic drug therapy. It might be possible to breastfeed safely during intermittent azacitidine therapy with an appropriate period of breastfeeding abstinence; the manufacturer recommends an abstinence period of 1 week after the last dose. Chemotherapy may adversely affect the normal microbiome and chemical makeup of breastmilk. Women who receive chemotherapy during pregnancy are more likely to have difficulty nursing their infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk A telephone follow-up study was conducted on 74 women who received cancer chemotherapy at one center during the second or third trimester of pregnancy to determine if they were successful at breastfeeding postpartum. Only 34% of the women were able to exclusively breastfeed their infants, and 66% of the women reported experiencing breastfeeding difficulties. This was in comparison to a 91% breastfeeding success rate in 22 other mothers diagnosed during pregnancy, but not treated with chemotherapy. Other statistically significant correlations included: 1. mothers with breastfeeding difficulties had an average of 5.5 cycles of chemotherapy compared with 3.8 cycles among mothers who had no difficulties; and 2. mothers with breastfeeding difficulties received their first cycle of chemotherapy on average 3.4 weeks earlier in pregnancy. Of the 9 women who received a fluorouracil-containing regimen, 8 had breastfeeding difficulties. Protein Binding Not available. |
||
| 参考文献 |
|
||
| 其他信息 |
Azacitidine can cause cancer according to The World Health Organization's International Agency for Research on Cancer (IARC).
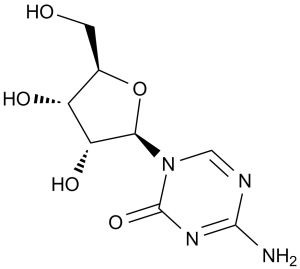
5-azacytidine is a white crystalline powder. (NTP, 1992) 5-azacytidine is an N-glycosyl-1,3,5-triazine that is 4-amino-1,3,5-triazin-2(1H)-one substituted by a beta-D-ribofuranosyl residue via an N-glycosidic linkage. An antineoplastic agent, it is used in the treatment of myeloid leukaemia. It has a role as an antineoplastic agent. It is a N-glycosyl-1,3,5-triazine and a nucleoside analogue. It is functionally related to a beta-D-ribose. Azacitidine is a pyrimidine nucleoside analogue with anti-neoplastic activity. It differs from cytosine by the presence of nitrogen in the C5-position, key in its hypomethylating activity. Two main mechanisms of action have been proposed for azacitidine. One of them is the induction of cytotoxicity. As an analogue of cytidine, it is able to incorporate into RNA and DNA, disrupting RNA metabolism and inhibiting protein and DNA synthesis. The other one is through the inhibition of DNA methyltransferase, impairing DNA methylation. Due to its anti-neoplastic activity and its ability to inhibit methylation in replicating DNA, azacytidine has been used mainly used in the treatment of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), two types of cancer characterized by the presence of aberrant DNA methylation. In May 2004, the FDA approved the use of azacitidine administered subcutaneously for the treatment of MDS of all French-American-British (FAB) subtypes. In January 2007, the FDA approved the intravenous administration of azacitidine. The use of oral azacitidine for the treatment of AML in patients in complete remission was approved by the FDA in September 2020. Azacitidine is a Nucleoside Metabolic Inhibitor. The mechanism of action of azacitidine is as a Nucleic Acid Synthesis Inhibitor. Azacitidine is a cytosine analogue and antineoplastic agent used in the therapy of myelodysplastic syndromes. Azacitidine is associated with a low rate of transient serum enzyme elevations during therapy and has not been convincingly implicated in cases of clinically apparent acute liver injury with jaundice. Azacitidine has been reported in Streptomyces sparsogenes with data available. Azacitidine is a pyrimidine nucleoside analogue of cytidine with antineoplastic activity. Azacitidine is incorporated into DNA, where it reversibly inhibits DNA methyltransferase, thereby blocking DNA methylation. Hypomethylation of DNA by azacitidine may activate tumor suppressor genes silenced by hypermethylation, resulting in an antitumor effect. This agent is also incorporated into RNA, thereby disrupting normal RNA function and impairing tRNA cytosine-5-methyltransferase activity. (NCI04) Azacitidine is only found in individuals that have used or taken this drug. It is a pyrimidine nucleoside analogue that inhibits DNA methyltransferase, impairing DNA methylation. It is also an antimetabolite of cytidine, incorporated primarily into RNA. Azacytidine has been used as an antineoplastic agent. Azacitidine (5-azacytidine) is a chemical analogue of the cytosine nucleoside used in DNA and RNA. Azacitidine is thought to induce antineoplastic activity via two mechanisms; inhibition of DNA methyltransferase at low doses, causing hypomethylation of DNA, and direct cytotoxicity in abnormal hematopoietic cells in the bone marrow through its incorporation into DNA and RNA at high doses, resulting in cell death. As azacitidine is a ribonucleoside, it incoporates into RNA to a larger extent than into DNA. The incorporation into RNA leads to the dissembly of polyribosomes, defective methylation and acceptor function of transfer RNA, and inhibition of the production of protein. Its incorporation into DNA leads to a covalent binding with DNA methyltransferases, which prevents DNA synthesis and subsequent cytotoxicity. A pyrimidine analogue that inhibits DNA methyltransferase, impairing DNA methylation. It is also an antimetabolite of cytidine, incorporated primarily into RNA. Azacytidine has been used as an antineoplastic agent. Drug Indication Azacitidine (for subcutaneous or intravenous use) is indicated for the treatment of adult patients with the following French-American-British (FAB) myelodysplastic syndrome (MDS) subtypes: refractory anemia (RA) or refractory anemia with ringed sideroblasts (RARS) (if accompanied by neutropenia or thrombocytopenia or requiring transfusions), refractory anemia with excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMMoL). Azacitidine is also indicated for the treatment of pediatric patients aged 1 month and older with newly diagnosed Juvenile Myelomonocytic Leukemia (JMML). Azacitidine (for oral use) is indicated for continued treatment of adult patients with acute myeloid leukemia (AML) who achieved first complete remission or complete remission with incomplete blood count recovery following intensive induction chemotherapy and are not able to complete intensive curative therapy. Azacitidine Mylan is indicated for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation (HSCT) with: intermediate 2 and high risk myelodysplastic syndromes (MDS) according to the International Prognostic Scoring System (IPSS),chronic myelomonocytic leukaemia (CMML) with 10 29% marrow blasts without myeloproliferative disorder,acute myeloid leukaemia (AML) with 20 30% blasts and multi lineage dysplasia, according to World Health Organisation (WHO) classification,AML with > 30% marrow blasts according to the WHO classification. Vidaza is indicated for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation (HSCT) with: Â intermediate 2 and high-risk myelodysplastic syndromes (MDS) according to the International Prognostic Scoring System (IPSS),chronic myelomonocytic leukaemia (CMML) with 10 29 % marrow blasts without myeloproliferative disorder,acute myeloid leukaemia (AML) with 20 30 % blasts and multi-lineage dysplasia, according to World Health Organisation (WHO) classification. Vidaza is indicated for the treatment of adult patients aged 65 years or older who are not eligible for HSCT with AML with > 30% marrow blasts according to the WHO classification. Azacitidine Accord is indicated for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation (HSCT) with: - intermediate-2 and high-risk myelodysplastic syndromes (MDS) according to the International Prognostic Scoring System (IPSS),- chronic myelomonocytic leukaemia (CMML) with 10-29 % marrow blasts without myeloproliferative disorder,- acute myeloid leukaemia (AML) with 20-30 % blasts and multi-lineage dysplasia, according to World Health Organisation (WHO) classification,- AML with > 30% marrow blasts according to the WHO classification. Azacitidine betapharm is indicated for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation (HSCT) with: intermediate-2 and high-risk myelodysplastic syndromes (MDS) according to the International Prognostic Scoring System (IPSS),chronic myelomonocytic leukaemia (CMML) with 10 % to 29 % marrow blasts without myeloproliferative disorder,acute myeloid leukaemia (AML) with 20 % to 30 % blasts and multi-lineage dysplasia, according to World Health Organization (WHO) classification,AML with > 30 % marrow blasts according to the WHO classification. Onureg is indicated as maintenance therapy in adult patients with acute myeloid leukaemia (AML) who achieved complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following induction therapy with or without consolidation treatment and who are not candidates for, including those who choose not to proceed to, hematopoietic stem cell transplantation (HSCT). Azacitidine Celgene is indicated for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation (HSCT) with: intermediate 2 and high-risk myelodysplastic syndromes (MDS) according to the International Prognostic Scoring System (IPSS),chronic myelomonocytic leukaemia (CMML) with 10 29 % marrow blasts without myeloproliferative disorder,acute myeloid leukaemia (AML) with 20 30 % blasts and multi-lineage dysplasia, according to World Health Organisation (WHO) classification,AML with > 30% marrow blasts according to the WHO classification. Treatment of myelodysplastic syndrome (including juvenile myelomonocytic leukaemia), Treatment of acute myeloid leukaemia Mechanism of Action Azacitidine (5-azacytidine) is a chemical analogue of the cytosine nucleoside present in DNA and RNA. It induces antineoplastic activity by inhibiting DNA methyltransferase at low doses and inducing cytotoxicity by incorporating itself into RNA and DNA at high doses. Covalent binding to DNA methyltransferase results in DNA hypomethylation and prevents DNA synthesis. On the other hand, the incorporation of azacitidine into RNA and DNA leads to cytotoxicity as follows: Following cellular uptake, azacitidine is phosphorylated by uridine-cytidine kinase to form 5-azacytidine monophosphate. Afterwards, pyrimidine monophosphate and diphosphate kinases phosphorylate 5-azacytidine monophosphate to form 5-azacytidine diphosphate and triphosphate, respectively. Azacitidine triphosphate is able to incorporate into RNA, disrupting RNA metabolism and protein synthesis. The reduction of azacytidine diphosphate leads to the formation of 5-aza-deoxycytidine diphosphate, which is then phosphorylated to form 5-azadeoxycitidine triphosphate, a compound able to incorporate into DNA and inhibit DNA synthesis. As a ribonucleoside, azacitidine incorporates into RNA to a larger extent than into DNA. Incorporating into RNA leads to the disassembly of polyribosomes, defective methylation and acceptor function of transfer RNA, and the inhibition of protein production, resulting in cell death. During the S-phase of the cell cycle, azacitidine exhibits the highest toxicity; however, the predominant mechanism of cytotoxicity has not been elucidated. The cytotoxic effects of azacitidine cause the death of rapidly dividing cells, including cancer cells that are no longer responsive to normal growth control mechanisms. Non-proliferating cells are relatively insensitive to azacitidine. It is believed that azacitidine exerts its antineoplastic effects through direct cytotoxicity on abnormal hematopoietic cells in the bone marrow. Telomerase activation is thought to be a critical step in cellular immortality and oncogenesis. Several reagents including differentiation-inducing and antineoplastic agents are known to inhibit telomerase activity, although the molecular mechanisms through which they inhibit telomerase activity remain unclear. Demethylating reagents have recently been used as potential antineoplastic drugs for some types of cancers including those of the prostate. In the present study, we examined the effect of the demethylating reagent 5-azacytidine (5-aza-CR) on telomerase activity using cells of two prostate cancer cell lines, DU-145 and TSU-PR1. 5-aza-CR treatment significantly reduced telomerase activity in TSU-PR1 cells, but not in DU-145 cells, although growth inhibition was observed to a similar extent in both cell lines. Reverse transcription-PCR analyses revealed that inhibition of telomerase activity was accompanied by down-regulation of telomerase catalytic subunit (hTERT) mRNA expression. Transient expression assays showed that 5-aza-CR repressed the transcriptional activity of the hTERT promoter and that the E-box within the core promoter was responsible for this down-regulation. Western blot analyses revealed that 5-aza-CR reactivated p16 expression and repressed c-Myc expression in TSU-PR1 cells but not in DU-145 cells. Overexpression of p16 in TSU-PR1 cells led to significant repression of c-Myc transcription. These findings suggest that 5-aza-CR inhibits telomerase activity via transcriptional repression of hTERT, in which p16 and c-Myc may play a key role. Cellular differentiation is controlled by a variety of factors including gene methylation, which represses particular genes as cell fate is determined. The incorporation of 5-azacytidine (5azaC) into DNA in vitro prevents methylation and thus can alter cellular differentiation pathways. Human bone marrow fibroblasts and MG63 cells treated with 5azaC were used as models of osteogenic progenitors and of a more mature osteoblast phenotype, respectively. The capacity for differentiation of these cells following treatment with glucocorticoids was investigated. 5azaC treatment led to significant expression of the osteoblastic marker alkaline phosphatase in MG63 osteosarcoma cells, which was further augmented by glucocorticoids; however, in human marrow fibroblasts alkaline phosphatase activity was only observed in glucocorticoid-treated cultures. MG63 cells represent a phenotype late in the osteogenic lineage in which demethylation is sufficient to induce alkaline phosphatase activity. Marrow fibroblasts are at an earlier stage of differentiation and require stimulation with glucocorticoids. In contrast, the expression of osteocalcin, an osteoblastic marker, was unaffected by 5azaC treatment, suggesting that regulation of expression of the osteocalcin gene does not involve methylation. These models provide novel approaches to the study of the control of differentiation in the marrow fibroblastic system. |
| 分子式 |
C8H12N4O5
|
|
|---|---|---|
| 分子量 |
244.2
|
|
| 精确质量 |
244.08
|
|
| 元素分析 |
C, 39.35; H, 4.95; N, 22.94; O, 32.76
|
|
| CAS号 |
320-67-2
|
|
| 相关CAS号 |
|
|
| PubChem CID |
9444
|
|
| 外观&性状 |
Crystals from methanol
|
|
| 密度 |
2.1±0.1 g/cm3
|
|
| 沸点 |
534.5±60.0 °C at 760 mmHg
|
|
| 熔点 |
226-232 °C (dec.)(lit.)
|
|
| 闪点 |
277.0±32.9 °C
|
|
| 蒸汽压 |
0.0±3.2 mmHg at 25°C
|
|
| 折射率 |
1.823
|
|
| LogP |
-1.99
|
|
| tPSA |
143.72
|
|
| 氢键供体(HBD)数目 |
4
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
17
|
|
| 分子复杂度/Complexity |
384
|
|
| 定义原子立体中心数目 |
4
|
|
| SMILES |
OC[C@H]1O[C@@H](N2C(N=C(N)N=C2)=O)[C@H](O)[C@@H]1O
|
|
| InChi Key |
NMUSYJAQQFHJEW-KVTDHHQDSA-N
|
|
| InChi Code |
InChI=1S/C8H12N4O5/c9-7-10-2-12(8(16)11-7)6-5(15)4(14)3(1-13)17-6/h2-6,13-15H,1H2,(H2,9,11,16)/t3-,4-,5-,6-/m1/s1
|
|
| 化学名 |
4-amino-1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1,3,5-triazin-2(1H)-one
|
|
| 别名 |
Vidaza; Abbreviations: 5AC; 5AZC. U 18496; U18496; 5-azacytidine; azacytidine; U-18496; ladakamycin. US brand names: Mylosar; Vidaza; 5-azacitidine;
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (8.52 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (8.52 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (8.52 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 30% propylene glycol, 5% Tween 80, 65% D5W:30mg/mL 配方 5 中的溶解度: 20 mg/mL (81.90 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0950 mL | 20.4750 mL | 40.9500 mL | |
| 5 mM | 0.8190 mL | 4.0950 mL | 8.1900 mL | |
| 10 mM | 0.4095 mL | 2.0475 mL | 4.0950 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT06190067 | Recruiting | Drug: Azacitidine Plus PD-1 therapy | Relapsed Classic Hodgkin Lymphoma Refractory Classic Hodgkin Lymphoma |
Navy General Hospital, Beijing | October 30, 2023 | Phase 2 |
| NCT03466294 | Active, not recruiting | Drug: Azacitidine and Venetoclax | Acute Myeloid Leukemia | University of Colorado, Denver | May 15, 2018 | Phase 2 |
| NCT04891068 | Recruiting | Drug: Azacitidine | Breast Cancer Female Breast Cancer Invasive |
University of Illinois at Chicago | January 10, 2022 | Phase 2 |
| NCT04187703 | Recruiting | Drug: 5-azacytidine Drug: Decitabine |
Myelodysplastic Syndromes MDS/MPN Crossover Syndromes |
Benjamin Tomlinson | November 16, 2020 | Early Phase 1 |
 DNMT3A-IN-3
DNMT3A-IN-3
 DNMT1-IN-3
DNMT1-IN-3
 BM30
BM30
 METTL16-IN-1
METTL16-IN-1
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
