| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| Other Sizes |
|
| 靶点 |
HMG-CoA reductase; HMG-CoA/3-hydroxy-3-methylglutaryl coenzyme A
|
|---|---|
| 体外研究 (In Vitro) |
阿托伐他汀治疗通过下调心肌梗死期间心肌细胞中 GRP78、caspase-12 和 CHOP 的表达,降低心肌细胞凋亡。此外,它还刺激内质网 (ER) 以应对心力衰竭和血管紧张素 II (Ang II) 刺激。 ) 紧张 [4]。
|
| 体内研究 (In Vivo) |
用电子压力计评价口服阿托伐他汀对小鼠脚爪炎性机械性高痛觉的影响。ELISA和RIA检测细胞因子和PGE(2)。 关键结果:在致敏动物中,阿托伐他汀治疗3天,剂量依赖性地减少了脂多糖(LPS)或抗原激发后引起的高痛觉。阿托伐他汀预处理可降低缓激肽和细胞因子(tnf - α、il -1 β和KC)诱导的高痛觉,以及脂多糖诱导的足跖皮肤il -1 β和PGE(2)的释放。在不影响血清胆固醇水平的情况下,甲羟戊酸联合治疗可阻止阿托伐他汀对lps诱导的高痛觉的抗痛觉作用。阿托伐他汀可抑制PGE诱导的高痛觉(2),提示阿托伐他汀的细胞内抗痛觉机制。阿托伐他汀对LPS或PGE(2)诱导的高痛觉的抗痛觉作用可以通过一氧化氮合酶(NOS)的非选择性抑制剂来阻止,但不能通过选择性抑制可诱导的NOS或缺乏这种酶的小鼠来阻止。[1]
|
| 细胞实验 |
细胞增殖测定基本上如前所述进行。简言之,将来自5名不同患者的SV-SMC以全生长培养基中每孔1×104个细胞的密度接种到24孔细胞培养板中。将细胞孵育过夜,然后在无血清培养基中静置3天,然后转移到含有5种不同浓度他汀类药物的全生长培养基(10%FCS)中。所有他汀类药物都在每个患者的细胞上进行了测试。2天后更换培养基和药物,4天后使用台盼蓝和血细胞仪在一式三个孔中测定活细胞数。细胞数的增加是通过从最终细胞数(第4天)中减去起始细胞数(0天)来计算的。然后将数据标准化为对照值(无他汀类药物),以校正来自不同患者的细胞之间增殖率的差异[2]。
|
| 动物实验 |
Effect of atorvastatin on hypernociception induced by LPS or antigen challenge [1]
To investigate the effect of atorvastatin on lipopolysaccharide (LPS)-induced inflammatory hypernociception, mice were pretreated orally with either atorvastatin, at doses of 1, 3, 10, 30 and 90 mg kg−1 or vehicle (PBS) once a day for 3 consecutive days. At 2 h after the last dose of atorvastatin, mice received an i.pl. injection of LPS (100 ng paw−1) or saline (vehicle for LPS). The animals were also treated with atorvastatin (30 mg kg−1) for 1 or 2 days before LPS challenge. The hypernociceptive responses were assessed 0.5, 1, 3, 5, 7 and 24 h after LPS or saline i.pl. injections. In addition, we investigated the effect of atorvastatin on the immune inflammatory hypernociception in mice sensitized to mBSA and challenged with antigen. The animals were pretreated orally with atorvastatin (30 mg kg−1) or PBS once a day for 3 consecutive days. At 2 h after the last dose of atorvastatin, mice received an i.pl. injection of mBSA (90 μg paw−1) or saline. In the control group, mBSA was injected into the paws of the false immunized mice (see above). Mice were fasted for 8 h receiving atorvastatin or PBS. The hypernociceptive responses were assessed 1, 3 and 5 h after challenge with antigen.
|
| 药代性质 (ADME/PK) |
Atorvastatin presents a dose-dependent and non-linear pharmacokinetic profile. It is very rapidly absorbed after oral administration. After the administration of a dose of 40 mg, its peak plasma concentration of 28 ng/ml is reached 1-2 hours after initial administration with an AUC of about 200 ng∙h/ml. Atorvastatin undergoes extensive first-pass metabolism in the wall of the gut and the liver, resulting in an absolute oral bioavailability of 14%. Plasma atorvastatin concentrations are lower (approximately 30% for Cmax and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration. Administration of atorvastatin with food results in prolonged Tmax and a reduction in Cmax and AUC. Breast Cancer Resistance Protein (BCRP) is a membrane-bound protein that plays an important role in the absorption of atorvastatin. Evidence from pharmacogenetic studies of c.421C>A single nucleotide polymorphisms (SNPs) in the gene for BCRP has demonstrated that individuals with the 421AA genotype have reduced functional activity and 1.72-fold higher AUC for atorvastatin compared to study individuals with the control 421CC genotype. This has important implications for the variation in response to the drug in terms of efficacy and toxicity, particularly as the BCRP c.421C>A polymorphism occurs more frequently in Asian populations than in Caucasians. Other statin drugs impacted by this polymorphism include [fluvastatin], [simvastatin], and [rosuvastatin]. Genetic differences in the OATP1B1 (organic-anion-transporting polypeptide 1B1) hepatic transporter encoded by the SCLCO1B1 gene (Solute Carrier Organic Anion Transporter family member 1B1) have been shown to impact atorvastatin pharmacokinetics. Evidence from pharmacogenetic studies of the c.521T>C single nucleotide polymorphism (SNP) in the gene encoding OATP1B1 (SLCO1B1) demonstrated that atorvastatin AUC was increased 2.45-fold for individuals homozygous for 521CC compared to homozygous 521TT individuals. Other statin drugs impacted by this polymorphism include [simvastatin], [pitavastatin], [rosuvastatin], and [pravastatin].
Route of Elimination Atorvastatin and its metabolites are mainly eliminated in the bile without enterohepatic recirculation. The renal elimination of atorvastatin is very minimal and represents less than 1% of the eliminated dose. Volume of Distribution The reported volume of distribution of atorvastatin is of 380 L. Clearance The registered total plasma clearance of atorvastatin is of 625 ml/min. /MILK/ In a separate experiment, a single dose of 10 mg/kg atorvastatin administered to female Wistar rats on gestation day 19 or lactation day 13 provided evidence of placental transfer and excretion into the milk. PMID:9520344 Lipitor and its metabolites are eliminated primarily in bile following hepatic and/or extra-hepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. ... Less than 2% of a dose of Lipitor is recovered in urine following oral administration. /MILK/ It is not known whether atorvastatin is excreted in human milk, but a small amount of another drug in this class does pass into breast milk. Nursing rat pups had plasma and liver drug levels of 50% and 40%, respectively, of that in their mother's milk. Mean volume of distribution of Lipitor is approximately 381 liters. Lipitor is >/= 98% bound to plasma proteins. A blood/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells. For more Absorption, Distribution and Excretion (Complete) data for ATORVASTATIN (8 total), please visit the HSDB record page. View More
Metabolism / Metabolites
Biological Half-Life The half-life of atorvastatin is 14 hours while the half-life of its metabolites can reach up to 30 hours. /MILK/ ...After administration to lactating rats, radioactivity in milk reached the maximum of 17.1 ng eq./mL at 6.0 hr and thereafter declined with a half-life of 7.8 hr. Nemoto H et al; Yakuri To Chiryo 26 (7): 79-96 (1998) Mean plasma elimination half-life of Lipitor in humans is approximately 14 hours, but the half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Atorvastatin is anticholesteremic agent and hydroxymethylglutaryl-CoA reductase inhibitor. HUMAN EXPOSURE AND TOXICITY: Cases of fatal and nonfatal hepatic failure have been reported rarely in patients receiving statins, including atorvastatin. Rhabdomyolysis with acute renal failure secondary to myoglobinuria also has been reported rarely in patients receiving statins, including atorvastatin. Lipid lowering drugs offer no benefit during pregnancy because cholesterol and cholesterol derivatives are needed for normal fetal development. Atherosclerosis is a chronic process, and discontinuation of lipid-lowering drugs during pregnancy should have little impact on long-term outcomes of primary hypercholesterolemia therapy. The occurrence of neuropsychiatric reactions is associated with statin treatment. They include behavioral alterations; cognitive and memory impairments; sleep disturbance; and sexual dysfunction. ANIMAL STUDIES: In a 2-year carcinogenicity study in rats at dose levels of 10, 30, and 100 mg/kg/day, 2 rare tumors were found in muscle in high-dose females: in one, there was a rhabdomyosarcoma, and in another, there was a fibrosarcoma. Atorvastatin caused no adverse effects on semen parameters, or reproductive organ histopathology in dogs given doses of 10, 40, or 120 mg/kg for two years. Male rats given 100 mg/kg/day for 11 weeks prior to mating had decreased sperm motility, spermatid head concentration, and increased abnormal sperm. Studies in rats performed at doses up to 175 mg/kg produced no changes in fertility. There was aplasia and aspermia in the epididymis of 2 of 10 rats treated with 100 mg/kg/day of atorvastatin for 3 months; testis weights were significantly lower at 30 and 100 mg/kg and epididymal weight was lower at 100 mg/kg. In a study in rats given 20, 100, or 225 mg/kg/day, from gestation day 7 through to lactation day 21 (weaning), there was decreased pup survival at birth, neonate, weaning, and maturity in pups of mothers dosed with 225 mg/kg/day. Body weight was decreased on days 4 and 21 in pups of mothers dosed at 100 mg/kg/day; pup body weight was decreased at birth and at days 4, 21, and 91 at 225 mg/kg/day. Pup development was delayed. In vitro, atorvastatin was not mutagenic or clastogenic in the following tests with and without metabolic activation: the Ames test with Salmonella typhimurium and Escherichia coli, the HGPRT forward mutation assay in Chinese hamster lung cells, and the chromosomal aberration assay in Chinese hamster lung cells. Atorvastatin was negative in the in vivo mouse micronucleus test. Atorvastatin selectively and competitively inhibits the hepatic enzyme HMG-CoA reductase. As HMG-CoA reductase is responsible for converting HMG-CoA to mevalonate in the cholesterol biosynthesis pathway, this results in a subsequent decrease in hepatic cholesterol levels. Decreased hepatic cholesterol levels stimulates upregulation of hepatic LDL-C receptors which increases hepatic uptake of LDL-C and reduces serum LDL-C concentrations. Hepatotoxicity Atorvastatin therapy is associated with mild, asymptomatic and usually transient serum aminotransferase elevations in 1% to 3% of patients but levels above 3 times ULN in less than 1%. In summary analyses of large scale studies with prospective monitoring, ALT elevations above 3 times the upper limit of normal (ULN) occurred in 0.7% of atorvastatin treated versus 0.3% of placebo recipients. These elevations were more common with higher doses of atorvastatin, being 2.3% with 80 mg daily. Most elevations were self-limited and did not require dose modification. Atorvastatin is also associated with frank, clinically apparent hepatic injury but this is rare, occurring in ~1:3000 to 1:5000 treated patients. The clinical presentation of atorvastatin hepatotoxicity varies greatly from simple cholestatic hepatitis, to mixed forms, to frankly hepatocellular injury. The latency to onset of injury is also highly variable ranging from 1 month to several years. However, most cases arise within 6 months of starting atorvastatin or several months after a dose escalation. The most common presentation is a cholestatic hepatitis that tends to be mild to moderate in severity and self-limiting in course (Cases 1 and 2). Atorvastatin hepatotoxicity can also present with a distinctly hepatocellular pattern of injury with marked elevations in serum aminotransferase levels and minimal or no increase in alkaline phosphatase. Rash, fever and eosinophilia are uncommon, but at least one-third of hepatocellular cases have features of autoimmunity, marked by high immunoglobulin levels, ANA positivity and liver biopsy findings of autoimmune hepatitis (Cases 3 and 4). These autoimmune cases usually resolve once atorvastatin is stopped, although they may require corticosteroid therapy for resolution. Strikingly, however, some cases of apparent autoimmune hepatitis caused by atorvastatin do not resolve with stopping the medication but are self-sustained and require long term immunosuppressive therapy. It is unclear whether these cases of persistent autoimmune hepatitis caused by the statin therapy or are triggered by statin in a susceptible host. Another possibility is that the association is coincidental and represents a de novo onset of autoimmune hepatitis in someone who happens to be taking a statin. Likelihood score: A (well known cause of clinically apparent liver injury). View More
Effects During Pregnancy and Lactation
Protein Binding Atorvastatin is highly bound to plasma proteins and over 98% of the administered dose is found in a bound form. |
| 参考文献 |
[1]. Santodomingo-Garzón T, et al. Atorvastatin inhibits inflammatory hypernociception. Br J Pharmacol. 2006 Sep;149(1):14-22.
[2]. Turner NA, et al. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007 Oct;50(4):458-61. [3]. Nawrocki, J.W., et al., Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler Thromb Vasc Biol, 1995. 15(5): p. 678-82. [4]. Song XJ, et al. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci. 2011;8(7):564-72. [5]. Li Y, et al. Inhibition of endoplasmic reticulum stress signaling pathway: A new mechanism of statins to suppress the development of abdominal aortic aneurysm. PLoS One. 2017 Apr 3;12(4):e0174821. [6]. Ming-Bai Hu, et al. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct Pathol. Sep-Oct 2018;42(5):409-415. [7]. In Vitro Screening for β-Hydroxy-β-methylglutaryl-CoA Reductase Inhibitory and Antioxidant Activity of Sequentially Extracted Fractions of Ficus palmata Forsk. Biomed Res Int. 2014; 2014: 762620. |
| 其他信息 |
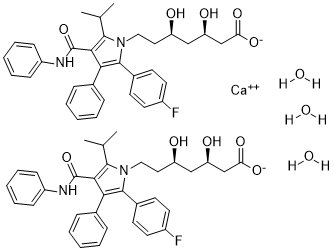
Atorvastatin calcium trihydrate is a hydrate that is the trihydrate form of atorvastatin calcium. It has a role as an environmental contaminant and a xenobiotic. It is a hydrate and a statin (synthetic). It contains an atorvastatin calcium.
Atorvastatin Calcium is the calcium salt of atorvastatin, a synthetic lipid-lowering agent. Atorvastatin competitively inhibits hepatic hydroxymethyl-glutaryl coenzyme A (HMG-CoA) reductase, the enzyme which catalyzes the conversion of HMG-CoA to mevalonate, a key step in cholesterol synthesis. This agent increases the number of LDL receptors on hepatic cell surfaces, enhancing the uptake and catabolism of LDL and reducing LDL production and the number of LDL particles, and lowers plasma cholesterol and lipoprotein levels. Like other statins, atorvastatin may also display direct antineoplastic activity, possibly by inhibiting farnesylation and geranylgeranylation of proteins such as small GTP-binding proteins, which may result in the arrest of cells in the G1 phase of the cell cycle. This agent may also sensitize tumor cells to cyctostatic drugs, possibly through the mTOR-dependent inhibition of Akt phosphorylation. A pyrrole and heptanoic acid derivative, HYDROXYMETHYLGLUTARYL-COA REDUCTASE INHIBITOR (statin), and ANTICHOLESTEREMIC AGENT that is used to reduce serum levels of LDL-CHOLESTEROL; APOLIPOPROTEIN B; and TRIGLYCERIDES. It is used to increase serum levels of HDL-CHOLESTEROL in the treatment of HYPERLIPIDEMIAS, and for the prevention of CARDIOVASCULAR DISEASES in patients with multiple risk factors. See also: Atorvastatin (has active moiety); Atorvastatin calcium trihydrate; ezetimibe (component of). Pure hypercholesterolaemia (heterozygous, homozygous, or otherwise primary hypercholesterolaemia), combined (mixed) hyperlipidaemia; prevention of cardiovascular events |
| 分子式 |
C66H74CAF2N4O13
|
|---|---|
| 分子量 |
1209.3876
|
| 精确质量 |
1208.484
|
| 元素分析 |
C, 65.55; H, 6.17; Ca, 3.31; F, 3.14; N, 4.63; O, 17.20
|
| CAS号 |
344423-98-9
|
| 相关CAS号 |
134523-03-8 (calcium);344423-98-9 (calcium trihydrate);134523-00-5 (free acid);134523-01-6 (sodium); 874114-41-7 (magnesium);
|
| PubChem CID |
656846
|
| 外观&性状 |
Typically exists as White to off-white solid at room temperature
|
| LogP |
9.91
|
| tPSA |
256.93
|
| 氢键供体(HBD)数目 |
9
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
22
|
| 重原子数目 |
86
|
| 分子复杂度/Complexity |
817
|
| 定义原子立体中心数目 |
4
|
| SMILES |
[Ca+2].FC1C([H])=C([H])C(=C([H])C=1[H])C1=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C(C(N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)=C(C([H])(C([H])([H])[H])C([H])([H])[H])N1C([H])([H])C([H])([H])[C@]([H])(C([H])([H])[C@]([H])(C([H])([H])C(=O)[O-])O[H])O[H].FC1C([H])=C([H])C(=C([H])C=1[H])C1=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C(C(N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)=C(C([H])(C([H])([H])[H])C([H])([H])[H])N1C([H])([H])C([H])([H])[C@]([H])(C([H])([H])[C@]([H])(C([H])([H])C(=O)[O-])O[H])O[H].O([H])[H].O([H])[H].O([H])[H]
|
| InChi Key |
SHZPNDRIDUBNMH-NIJVSVLQSA-L
|
| InChi Code |
InChI=1S/2C33H35FN2O5.Ca.3H2O/c2*1-21(2)31-30(33(41)35-25-11-7-4-8-12-25)29(22-9-5-3-6-10-22)32(23-13-15-24(34)16-14-23)36(31)18-17-26(37)19-27(38)20-28(39)40/h2*3-16,21,26-27,37-38H,17-20H2,1-2H3,(H,35,41)(H,39,40)3*1H2/q+2/p-2/t2*26-,27-/m11..../s1
|
| 化学名 |
calcium
(3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate
trihydrate
|
| 别名 |
liptonorm; CI-981; CI 981; CI981; Atorvastatin; atorvastatin calcium trihydrate; atorvastatin calcium trihydrate; 344423-98-9; Atorvastatin hemicalcium trihydrate; Totalip; Atorvastatin calcium salt trihydrate; ATORVASTATIN CALCIUM; Torvast; Atorvastatin calcium [USAN]; atorvastatin calcium salt
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.8269 mL | 4.1343 mL | 8.2686 mL | |
| 5 mM | 0.1654 mL | 0.8269 mL | 1.6537 mL | |
| 10 mM | 0.0827 mL | 0.4134 mL | 0.8269 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1