| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| 50g |
|
||
| Other Sizes |
|
| 靶点 |
Beta-1 adrenergic receptors
|
|---|---|
| 体外研究 (In Vitro) |
研究了硝苯地平和阿替洛尔单独或联合对大鼠主动脉平滑肌细胞增殖和迁移的体外影响。硝苯地平在10至100微M的浓度范围内抑制动脉肌细胞的复制。以细胞数量评估的抑制作用是剂量和时间依赖性的,48小时和72小时后的IC50分别为39和34微M;细胞倍增时间随着药物浓度的增加而增加,最高可达118小时,而对照组为28小时。单独使用阿替洛尔不能减少动脉肌细胞增殖,也不影响硝苯地平对细胞增殖的影响。硝苯地平和阿替洛尔单独以剂量依赖的方式抑制了纤维蛋白原作为趋化剂诱导的大鼠主动脉肌细胞迁移。当研究硝苯地平-阿替洛尔的组合时,观察到对细胞迁移的加性抑制作用。这些结果为这种药物联合对动脉粥样硬化早期步骤的潜在影响提供了体外支持。[3]
用普萘洛尔、阿替洛尔或美托洛尔治疗的血红素EC显示阳性LysoTracker红染色。β受体阻滞剂治疗的Hem-ECs中记录了LC3BII/LC3BI比值的增加以及p62的调节。在体外暴露于β-阻断剂的培养Hem-ECs中,大量的自噬空泡和多层体表征了自噬的细胞质超微结构特征。重要的是,在雷帕霉素治疗后观察到类似的自噬生化和形态学证据,而巴非霉素A1显著阻止了β受体阻滞剂在Hem-ECs中促进的自噬通量。 结论:我们的数据表明,自噬可能是β受体阻滞剂的作用机制之一,这为这类药物在涉及不受控制的血管生成的病理条件下的潜在治疗应用提供了新的机制见解[4]。 |
| 体内研究 (In Vivo) |
与用药前的血压相比,除了低剂量的舒张压外,所有3个剂量的阿替洛尔和氨氯地平联合用药在给药后24小时均显著降低了血压。与对照组相比,所有3剂阿替洛尔和氨氯地平联合用药均显著降低了给药后24小时的平均血压水平;此外,高剂量和中等剂量也显著降低了同期的BPV水平。通过概率和分析计算出的给药后24小时内收缩压和舒张压的q值分别为2.29和1.45,同期收缩压和舒张压分别为1.41和1.60。
结论:阿替洛尔和氨氯地平在降低和稳定2K1C肾血管性高血压大鼠血压方面具有显著的协同作用[5]。
|
| 细胞实验 |
对7名患有增殖性IH的儿童手术切除的新鲜组织标本进行酶消化。用抗人CD31免疫标记的磁性微珠分选细胞。根据表型特征,在P2至P6时,将扩增的Hem-ECs单独暴露于不同浓度(50μM至150μM)的普萘洛尔、阿替洛尔或美托洛尔,并与自噬抑制剂Bafilomycin A1联合使用。雷帕霉素是一种强效的自噬诱导剂,也被用作对照。通过Lysotracker Red染色、LC3BII/LC3BI和p62的蛋白质印迹分析以及透射电子显微镜的形态学分析来评估自噬[4]。
|
| 动物实验 |
Aim: To test the synergistic effects of atenolol and amlodipine on lowering blood pressure (BP) and reducing blood pressure variability (BPV) in 2-kidney, one-clip (2K1C) renovascular hypertensive rats.
Methods: Forty-eight 2K1C renovascular hypertensive rats were randomly divided into 6 groups. They were respectively given 0.8% carboxymethylcellulose sodium (control), atenolol (10.0 mg/kg), amlodipine (1.0 mg/kg), and combined atenolol and amlodipine (low dose: 5.0+0.5 mg/kg; intermediate dose: 10.0+1.0 mg/kg; high dose: 20.0+2.0 mg/kg). The drugs were given via a catheter in a gastric fistula. BP was recorded for 25 h from 1 h before drug administration to 24 h after administration.[5]
Animals and RVHR preparation [5] Male Sprague–Dawley rats (160–180 g) were anesthetized with a combination of ketamine (40 mg/kg) and diazepam (6 mg/kg). The right renal artery of each animal was isolated through a flank incision as described previously, and a silver clip (0.2 mm internal gap) was placed on the renal artery. Five weeks after placement of the clip, the systolic blood pressure (SBP) of rats was measured by using the tail-cuff method (CB10). In total, 48 RVHR whose SBP was greater than 160 mmHg were used in this study. Rats were kept in a controlled temperature (23 °C– 25 °C) and lighting (light 08:00–20:00, dark 20:00–08:00) environment, and had free access to food and tap water. All the animals used in this work received humane care in compliance with institutional animal care guidelines. BP and BPV measurement [5] SBP, diastolic blood pressure (DBP) and heart period (HP) were continuously recorded using a previously described technique. Briefly, rats were anesthetized by injection (ip) with a combination of ketamine (40 mg/kg) and diazepam (6 mg/kg). A floating polyethylene catheter was inserted into the lower abdominal aorta via the left femoral artery for BP measurement, and another catheter was placed into the stomach via a mid-abdominal incision for drug administration. The catheters were exteriorized through the interscapular skin. After a 2-d recovery period, the animals were placed for BP recording in individual cylindrical cages containing food and water. The aortic catheter was connected to a BP transducer via a rotating swivel that allowed the animals to move freely in the cage. After approximately 14-h habituation, at 9:00 o’clock the BP signal was begun to be digitized by a microcomputer. One hour later, at 10:00 o’clock the drug was given via the catheter in the gastric fistula. SBP, DBP, and HP values were recorded beat-to-beat for 25 h, up to 10:00 o’clock on the second day. The mean values of these parameters during a designated period were calculated and served as SBP, DBP and HP values. The standard deviation of all values obtained over 24 h was denoted as the quantitative parameter of variability; that is, SBP variability (SBPV), DBP variability (DBPV), and HP variability (HPV) for each rat. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Approximately 50% of an oral dose is absorbed from the gastrointestinal tract, with the remainder being excreted unchanged in the feces. Administering atenolol with food can decrease the AUC by about 20%. While atenolol can cross the blood-brain barrier, it does so slowly and to a small extent. 85% is eliminated by the kidneys following IV administration with 10% appearing in the feces. Total Vd of 63.8-112.5 L. Atenolol distributes into a central volume of 12.8-17.5 L along with two peripheral compartments with a combined volume of 51-95 L. Distribution takes about 3 hrs for the central compartment, 4 hrs for the shallower peripheral compartment, and 5-6 hrs for the deeper peripheral compartment. Total clearance is estimated at 97.3-176.3 mL/min with a renal clearance of 95-168 mL/min. In animals, atenolol is well distributed into most tissues and fluids except brain and /cerebrospinal fluid/. Unlike propranolol, only a small portion of atenolol is apparently distributed into the CNS. Approximately 5-15% of atenolol is bound to plasma protein. Atenolol readily crosses the placenta, and has been detected in cord blood. During continuous administration, fetal serum concentrations of the drug are probably equivalent to those in maternal serum. Atenolol is distributed into milk; peak milk concentrations of the drug are higher than peak serum concentrations after an individual dose, and the area under the milk concentration-time (AUC) is substantially greater than that of the serum AUC in lactating women receiving the drug continuously. Atenolol is rapidly but incompletely absorbed from the GI tract. Only about 50-60% of an oral dose of atenolol is absorbed. In healthy adults, peak plasma concentrations of 1-2 ug/ml are achieved 2-4 hours after oral administration of a single 200 mg dose of atenolol. An approximately fourfold interindividual variation in plasma concentrations attained has been reported with a specific oral dose of atenolol. Peak plasma atenolol concentrations are achieved within 5 minutes following direct IV injection of the drug, and decline rapidly during an initial distribution phase; after the first 7 hours, plasma concentrations reportedly decline with an elimination half-life similar to that of orally administered drug. For more Absorption, Distribution and Excretion (Complete) data for ATENOLOL (6 total), please visit the HSDB record page. Metabolism / Metabolites Minimal metabolism in the liver. The sole non-conjugated metabolite is the product of a hydroxylation reaction at the carbon between the amide and benzene groups. The only other metabolite to be confirmed is a glucuronide conjugate. These metabolites make up 5-8% and 2% of the renally excreted dose with 87-90% appearing as unchanged drug. The hydroxylated metabolite is exerts 1/10th the beta-blocking activity of atenolol. Minimal hepatic metabolism; removable by hemodialysis; very low lipid solubility. Little or no metabolism of atenolol occurs in the liver. Approximately 40-50% of an oral dose of the drug is excreted in urine unchanged. The remainder is excreted unchanged in feces, principally as unabsorbed drug. About 1-12% of atenolol is reportedly removed by hemodialysis. Hepatic (minimal) Route of Elimination: Approximately 50% of an oral dose is absorbed from the gastrointestinal tract, the remainder being excreted unchanged in the feces. Unlike propranolol or metoprolol, but like nadolol, atenolol undergoes little or no metabolism by the liver, and the absorbed portion is eliminated primarily by renal excretion. Half Life: 6-7 hours Biological Half-Life 6-7 hrs. In patients with normal renal function, atenolol has a plasma half-life (t1/2) of 6-7 hours. Children with normal renal function may exhibit a shorter elimination half-life. In one study in children ages 5-16 (mean: 8.9) with arhythmias and normal renal and hepatic function, the terminal elimination half-life averaged 4.6 hours. Plasma t1/2 of the drug increases to 16-27 hours in patients with creatinine clearances of 15-35 ml/minute per 1.73 sq m and exceeds 27 hours with progressive renal impairment. The half-life in the elderly was significantly longer (8.8 + or - 0.9 hr) compared with that in the young (5.8 + or - 1.1 hr) (p < 0.01). |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
Like metoprolol, atenolol competes with sympathomimetic neurotransmitters such as catecholamines for binding at beta(1)-adrenergic receptors in the heart and vascular smooth muscle, inhibiting sympathetic stimulation. This results in a reduction in resting heart rate, cardiac output, systolic and diastolic blood pressure, and reflex orthostatic hypotension. Higher doses of atenolol also competitively block beta(2)-adrenergic responses in the bronchial and vascular smooth muscles. Toxicity Data LD50: 2000-3000 mg/kg(oral, mice). Interactions Concomitant administration of atenolol with reserpine may increase the incidence of hypotension and bradycardia as compared with atenolol alone, because of reserpine's catecholamine-depleting activity. Atenolol also is additive with and may potentiate the hypotensive actions of other hypotensive agents (e.g., hydralazine, methylodpa). It has been shown that paradoxical pressor response to a beta adrenoceptor antagonist occurs in conscious rats pretreated with an alpha adrenoceptor antagonist. This study examines the influence of anaesthetic agents on mean arterial pressure response to a beta blocker. ... IV injections of all three beta blockers caused dose dependent increases in mean arterial pressure in urethane anesthetized rats. In halothane anesthetized rats, propranolol and atenolol did not alter mean arterial pressure ... In the presence of pentobarbitone, none of the beta blockers raised mean arterial pressure. ... /When/ propranolol or atenolol were iv injected in pentobarbitone anesthetized rats treated with both phentolamine and adrenaline, /they both/ ... raised the mean arterial pressure. /These/ results show that anesthetic agents differentially affect the mean arterial pressure response to a beta blocker. In a recent study in which metabolic characteristics of newly detected obese and nonobese hypertensive subjects were compared with those of normotensive subjects, insulin sensitivity was decreased, fasting insulin values and insulin values after an intravenous glucose tolerance test were increased, and fasting and intravenous glucose tolerance glucose values were increased in both hypertensive groups. ... The effects of various antihypertensive agents on these metabolic variables have been assessed in prospective trials. Treatment with the beta 1 selective blocking agents metoprolol and atenolol was associated with decreased insulin sensitivity and increased fasting values of insulin and glucose. A double blind, randomized, crossover study of the pharmacokinetics of nifedipine and atenolol was conducted in 15 healthy male subjects (mean age 32 yr) who received a single oral tablet containing 50 mg atenolol, a sustained action tablet containing 20 mg nifedipine, both the atenolol and nifedipine tablets together, or an oral capsule containing 50 mg atenolol and 20 mg sustained release nifedipine. There was no difference between atenolol alone, given with nifedipine tablet or in the combination tablet with respect to time to maximum blood concentration or elimination half-life. ... /In conclusion,/ the fixed combination of nifedipine and atenolol is bioequivalent to the free combination and the bioavailability of both drugs in the fixed combination is equivalent to that of the single tablets. For more Interactions (Complete) data for ATENOLOL (14 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Mouse oral 2000 mg/kg LD50 Rat oral 3000 mg/kg LD50 Mouse iv 98.7 mg/kg LD50 Rat iv 59.24 mg/kg |
| 参考文献 |
|
| 其他信息 |
Therapeutic Uses
Adrenergic beta-Antagonists; Anti-Arrhythmia Agents; Antihypertensive Agents; Sympatholytics Atenolol has been used with good results alone or in conjunction with a benzodiazepine in the management of acute alcohol withdrawal in a limited number of patients. Atenolol ... /is/ indicated in the treatment of classic angina pectoris, also referred to as "effort-associated angina". /Included in US product labeling/ Atenolol /is/ used in the treatment of mitral value prolapse syndrome. /NOT included in US product labeling/ For more Therapeutic Uses (Complete) data for ATENOLOL (12 total), please visit the HSDB record page. Drug Warnings Atenolol should be used with caution and in reduced dosage in patients with impaired renal function, especially when creatinine clearance is less than 35 ml/minute per 1.73 sq m. ... Patients receiving atenolol after hemodialysis /should/ be administered the drug under close supervision in a hospital setting, since marked hypotension may occur. Atenolol is contraindicated in patients with sinus bradycardia, AV block greater than first degree, cardiogenic shock, and overt cardiac failure. Atenolol should be used with caution in patients undergoing major surgery involving general anesthesia. The necessity of withdrawing beta-adrenergic blocking therapy prior to major surgery is controversial. Severe, protracted hypotension and difficulty in restarting or maintaining a heart beat have occurred during surgery in some patients who have received beta-adrenergic blocking agents. Abrupt withdrawal of atenolol may exacerbate angina symptoms and/or precipitate myocardial infarction and venticular arrhythmias in patients with coronary artery disease, or may precipitate thyroid storm in patients with thyrotoxicosis. Therefore, patients receiving atenolol (especially those with ischemic heart disease) should be warned not to interrupt or discontinue therapy without consulting their physician. For more Drug Warnings (Complete) data for ATENOLOL (12 total), please visit the HSDB record page. Pharmacodynamics Atenolol is a cardio-selective beta-blocker and as such exerts most of its effects on the heart. It acts as an antagonist to sympathetic innervation and prevents increases in heart rate, electrical conductivity, and contractility in the heart due to increased release of norepinephrine from the peripheral nervous system. Together the decreases in contractility and rate produce a reduction in cardiac output resulting in a compensatory increase in peripheral vascular resistance in the short-term. This response later declines to baseline with long-term use of atenolol. More importantly, this reduction in the work demanded of the myocardium also reduces oxygen demand which provides therapeutic benefit by reducing the mismatch of oxygen supply and demand in settings where coronary blood flow is limited, such as in coronary atherosclerosis. Reducing oxygen demand, particularly due to exercise, can reduce the frequency of angina pectoris symptoms and potentially improve survival of the remaining myocardium after myocardial infarction. The decrease in rate of sinoatrial node potentials, electrical conduction, slowing of potentials traveling through the atrioventricular node, and reduced frequency of ectopic potentials due to blockade of adrenergic beta receptors has led to benefit in arrhythmic conditions such as atrial fibrillation by controlling the rate of action potential generation and allowing for more effective coordinated contractions. Since a degree of sympathetic activity is necessary to maintain cardiac function, the reduced contractility induced by atenolol may precipitate or worsen heart failure, especially during volume overload. The effects of atenolol on blood pressure have been established, although it is less effective than alternative beta-blockers, but the mechanism has not yet been characterized. As a β1 selective drug, it does not act via the vasodilation produced by non-selective agents. Despite this there is a sustained reduction in peripheral vascular resistance, and consequently blood pressure, alongside a decrease in cardiac output. It is thought that atenolol's antihypertensive activity may be related to action on the central nervous system (CNS) or it's inhibition of the renin-aldosterone-angiotensin system rather than direct effects on the vasculature. Atenolol produces CNS effects similar to other beta-blockers, but does so to a lesser extent due to reduces ability to cross the blood-brain barrier. It has the potential to produce fatigue, depression, and sleep disturbances such as nightmares or insomnia. The exact mechanisms behind these have not been characterized but their occurrence must be considered as they represent clinically relevant adverse effects. Atenolol exerts some effects on the respiratory system although to a much lesser extent than non-selective beta-blockers. Interaction with β2 receptors in the airways can produce bronchoconstriction by blocking the relaxation of bronchial smooth muscle mediated by the sympathetic nervous system. The same action can interfere with β-agonist therapies used in asthma and chronic obstructive pulmonary disease. Unlike some other beta-blocker drugs, atenolol does not have intrinsic sympathomimetic or membrane stabilizing activity nor does it produce changes in glycemic control. |
| 分子式 |
C14H22N2O3
|
|---|---|
| 分子量 |
266.34
|
| 精确质量 |
266.163
|
| 元素分析 |
C, 63.13; H, 8.33; N, 10.52; O, 18.02
|
| CAS号 |
29122-68-7
|
| 相关CAS号 |
Atenolol-d7;1202864-50-3; 51706-40-2 (HCl); 29122-68-7;93379-54-5 (S isomer); 56715-13-0 (R isomer)
|
| PubChem CID |
2249
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
508.0±50.0 °C at 760 mmHg
|
| 熔点 |
154°C
|
| 闪点 |
261.1±30.1 °C
|
| 蒸汽压 |
0.0±1.4 mmHg at 25°C
|
| 折射率 |
1.540
|
| LogP |
0.1
|
| tPSA |
84.58
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
19
|
| 分子复杂度/Complexity |
263
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O(C1C([H])=C([H])C(C([H])([H])C(N([H])[H])=O)=C([H])C=1[H])C([H])([H])C([H])(C([H])([H])N([H])C([H])(C([H])([H])[H])C([H])([H])[H])O[H]
|
| InChi Key |
METKIMKYRPQLGS-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C14H22N2O3/c1-10(2)16-8-12(17)9-19-13-5-3-11(4-6-13)7-14(15)18/h3-6,10,12,16-17H,7-9H2,1-2H3,(H2,15,18)
|
| 化学名 |
2-[4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]acetamide
|
| 别名 |
Atenolol; Blokium; Normiten; atenolol; 29122-68-7; Prenormine; Blokium; Myocord; Normiten; (RS)-Atenolol; Tenormine; Tenormin
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~375.46 mM)
H2O : ~8.33 mg/mL (~31.28 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 36.67 mg/mL (137.68 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.7546 mL | 18.7730 mL | 37.5460 mL | |
| 5 mM | 0.7509 mL | 3.7546 mL | 7.5092 mL | |
| 10 mM | 0.3755 mL | 1.8773 mL | 3.7546 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04931108 | RECRUITING | Drug: Nitrendipine/Atenolol Drug: Nitrendipine Drug: Atenolol |
Hypertension | Shanghai Jiao Tong University School of Medicine | 2021-09-27 | Phase 4 |
| NCT00913965 | COMPLETED | Drug: Atenolol Tablets 100 mg (Cord Laboratories) Drug: Atenolol Tablets 100 mg (Stuart Pharmaceutical) |
Hypertension | Sandoz | 1989-07 | Phase 1 |
| NCT01719367 | COMPLETEDWITH RESULTS | Drug: Atenolol | Atrial Fibrillation | Vanderbilt University Medical Center | 2013-01 | Not Applicable |
| NCT01397994 | UNKNOWN STATUS | Drug: Nicorandil Drug: Atenolol |
Chronic Stable Angina | Ferozsons Laboratories Ltd. | 2011-09 | Phase 4 |
| NCT04905277 | ACTIVE, NOT RECRUITING | Drug: Atenolol 50 MG Drug: Placebo |
Healthy | Sundeep Khosla, M.D. | 2021-07-27 | Phase 2 |
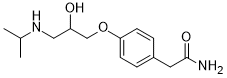
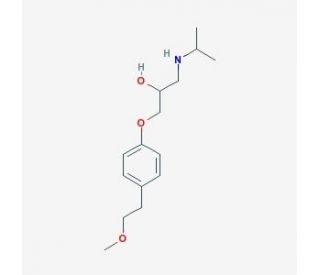 美托洛尔
美托洛尔
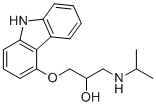 咔唑心安
咔唑心安
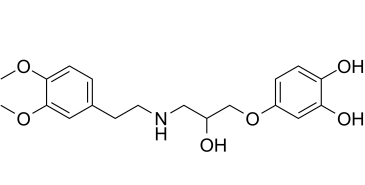 Ro 363
Ro 363
 DSP-4 hydrochloride
DSP-4 hydrochloride
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























