| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
Beta-1 adrenergic receptors
|
|---|---|
| 体外研究 (In Vitro) |
研究了硝苯地平和阿替洛尔单独或联合对大鼠主动脉平滑肌细胞增殖和迁移的体外影响。硝苯地平在10至100微M的浓度范围内抑制动脉肌细胞的复制。以细胞数量评估的抑制作用是剂量和时间依赖性的,48小时和72小时后的IC50分别为39和34微M;细胞倍增时间随着药物浓度的增加而增加,最高可达118小时,而对照组为28小时。单独使用阿替洛尔不能减少动脉肌细胞增殖,也不影响硝苯地平对细胞增殖的影响。硝苯地平和阿替洛尔单独以剂量依赖的方式抑制了纤维蛋白原作为趋化剂诱导的大鼠主动脉肌细胞迁移。当研究硝苯地平-阿替洛尔的组合时,观察到对细胞迁移的加性抑制作用。这些结果为这种药物联合对动脉粥样硬化早期步骤的潜在影响提供了体外支持。[3]
用普萘洛尔、阿替洛尔或美托洛尔治疗的血红素EC显示阳性LysoTracker红染色。β受体阻滞剂治疗的Hem-ECs中记录了LC3BII/LC3BI比值的增加以及p62的调节。在体外暴露于β-阻断剂的培养Hem-ECs中,大量的自噬空泡和多层体表征了自噬的细胞质超微结构特征。重要的是,在雷帕霉素治疗后观察到类似的自噬生化和形态学证据,而巴非霉素A1显著阻止了β受体阻滞剂在Hem-ECs中促进的自噬通量。 结论:我们的数据表明,自噬可能是β受体阻滞剂的作用机制之一,这为这类药物在涉及不受控制的血管生成的病理条件下的潜在治疗应用提供了新的机制见解[4]。 |
| 体内研究 (In Vivo) |
与用药前的血压相比,除了低剂量的舒张压外,所有3个剂量的阿替洛尔和氨氯地平联合用药在给药后24小时均显著降低了血压。与对照组相比,所有3剂阿替洛尔和氨氯地平联合用药均显著降低了给药后24小时的平均血压水平;此外,高剂量和中等剂量也显著降低了同期的BPV水平。通过概率和分析计算出的给药后24小时内收缩压和舒张压的q值分别为2.29和1.45,同期收缩压和舒张压分别为1.41和1.60。 结论:阿替洛尔和氨氯地平在降低和稳定2K1C肾血管性高血压大鼠血压方面具有显著的协同作用[5]。
|
| 细胞实验 |
对7名患有增殖性IH的儿童手术切除的新鲜组织标本进行酶消化。用抗人CD31免疫标记的磁性微珠分选细胞。根据表型特征,在P2至P6时,将扩增的Hem-ECs单独暴露于不同浓度(50μM至150μM)的普萘洛尔、阿替洛尔或美托洛尔,并与自噬抑制剂Bafilomycin A1联合使用。雷帕霉素是一种强效的自噬诱导剂,也被用作对照。通过Lysotracker Red染色、LC3BII/LC3BI和p62的蛋白质印迹分析以及透射电子显微镜的形态学分析来评估自噬[4]。
|
| 动物实验 |
Aim: To test the synergistic effects of atenolol and amlodipine on lowering blood pressure (BP) and reducing blood pressure variability (BPV) in 2-kidney, one-clip (2K1C) renovascular hypertensive rats.
Methods: Forty-eight 2K1C renovascular hypertensive rats were randomly divided into 6 groups. They were respectively given 0.8% carboxymethylcellulose sodium (control), atenolol (10.0 mg/kg), amlodipine (1.0 mg/kg), and combined atenolol and amlodipine (low dose: 5.0+0.5 mg/kg; intermediate dose: 10.0+1.0 mg/kg; high dose: 20.0+2.0 mg/kg). The drugs were given via a catheter in a gastric fistula. BP was recorded for 25 h from 1 h before drug administration to 24 h after administration.[5]
Animals and RVHR preparation [5] Male Sprague–Dawley rats (160–180 g) were anesthetized with a combination of ketamine (40 mg/kg) and diazepam (6 mg/kg). The right renal artery of each animal was isolated through a flank incision as described previously, and a silver clip (0.2 mm internal gap) was placed on the renal artery. Five weeks after placement of the clip, the systolic blood pressure (SBP) of rats was measured by using the tail-cuff method (CB10). In total, 48 RVHR whose SBP was greater than 160 mmHg were used in this study. Rats were kept in a controlled temperature (23 °C– 25 °C) and lighting (light 08:00–20:00, dark 20:00–08:00) environment, and had free access to food and tap water. All the animals used in this work received humane care in compliance with institutional animal care guidelines. BP and BPV measurement [5] SBP, diastolic blood pressure (DBP) and heart period (HP) were continuously recorded using a previously described technique. Briefly, rats were anesthetized by injection (ip) with a combination of ketamine (40 mg/kg) and diazepam (6 mg/kg). A floating polyethylene catheter was inserted into the lower abdominal aorta via the left femoral artery for BP measurement, and another catheter was placed into the stomach via a mid-abdominal incision for drug administration. The catheters were exteriorized through the interscapular skin. After a 2-d recovery period, the animals were placed for BP recording in individual cylindrical cages containing food and water. The aortic catheter was connected to a BP transducer via a rotating swivel that allowed the animals to move freely in the cage. After approximately 14-h habituation, at 9:00 o’clock the BP signal was begun to be digitized by a microcomputer. One hour later, at 10:00 o’clock the drug was given via the catheter in the gastric fistula. SBP, DBP, and HP values were recorded beat-to-beat for 25 h, up to 10:00 o’clock on the second day. The mean values of these parameters during a designated period were calculated and served as SBP, DBP and HP values. The standard deviation of all values obtained over 24 h was denoted as the quantitative parameter of variability; that is, SBP variability (SBPV), DBP variability (DBPV), and HP variability (HPV) for each rat. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Approximately 50% of an oral dose is absorbed from the gastrointestinal tract, with the remainder being excreted unchanged in the feces. Administering atenolol with food can decrease the AUC by about 20%. While atenolol can cross the blood-brain barrier, it does so slowly and to a small extent. 85% is eliminated by the kidneys following IV administration with 10% appearing in the feces. Total Vd of 63.8-112.5 L. Atenolol distributes into a central volume of 12.8-17.5 L along with two peripheral compartments with a combined volume of 51-95 L. Distribution takes about 3 hrs for the central compartment, 4 hrs for the shallower peripheral compartment, and 5-6 hrs for the deeper peripheral compartment. Total clearance is estimated at 97.3-176.3 mL/min with a renal clearance of 95-168 mL/min. In animals, atenolol is well distributed into most tissues and fluids except brain and /cerebrospinal fluid/. Unlike propranolol, only a small portion of atenolol is apparently distributed into the CNS. Approximately 5-15% of atenolol is bound to plasma protein. Atenolol readily crosses the placenta, and has been detected in cord blood. During continuous administration, fetal serum concentrations of the drug are probably equivalent to those in maternal serum. Atenolol is distributed into milk; peak milk concentrations of the drug are higher than peak serum concentrations after an individual dose, and the area under the milk concentration-time (AUC) is substantially greater than that of the serum AUC in lactating women receiving the drug continuously. Atenolol is rapidly but incompletely absorbed from the GI tract. Only about 50-60% of an oral dose of atenolol is absorbed. In healthy adults, peak plasma concentrations of 1-2 ug/ml are achieved 2-4 hours after oral administration of a single 200 mg dose of atenolol. An approximately fourfold interindividual variation in plasma concentrations attained has been reported with a specific oral dose of atenolol. Peak plasma atenolol concentrations are achieved within 5 minutes following direct IV injection of the drug, and decline rapidly during an initial distribution phase; after the first 7 hours, plasma concentrations reportedly decline with an elimination half-life similar to that of orally administered drug. For more Absorption, Distribution and Excretion (Complete) data for ATENOLOL (6 total), please visit the HSDB record page. Metabolism / Metabolites Minimal metabolism in the liver. The sole non-conjugated metabolite is the product of a hydroxylation reaction at the carbon between the amide and benzene groups. The only other metabolite to be confirmed is a glucuronide conjugate. These metabolites make up 5-8% and 2% of the renally excreted dose with 87-90% appearing as unchanged drug. The hydroxylated metabolite is exerts 1/10th the beta-blocking activity of atenolol. Minimal hepatic metabolism; removable by hemodialysis; very low lipid solubility. Little or no metabolism of atenolol occurs in the liver. Approximately 40-50% of an oral dose of the drug is excreted in urine unchanged. The remainder is excreted unchanged in feces, principally as unabsorbed drug. About 1-12% of atenolol is reportedly removed by hemodialysis. Hepatic (minimal) Route of Elimination: Approximately 50% of an oral dose is absorbed from the gastrointestinal tract, the remainder being excreted unchanged in the feces. Unlike propranolol or metoprolol, but like nadolol, atenolol undergoes little or no metabolism by the liver, and the absorbed portion is eliminated primarily by renal excretion. Half Life: 6-7 hours Biological Half-Life 6-7 hrs. In patients with normal renal function, atenolol has a plasma half-life (t1/2) of 6-7 hours. Children with normal renal function may exhibit a shorter elimination half-life. In one study in children ages 5-16 (mean: 8.9) with arhythmias and normal renal and hepatic function, the terminal elimination half-life averaged 4.6 hours. Plasma t1/2 of the drug increases to 16-27 hours in patients with creatinine clearances of 15-35 ml/minute per 1.73 sq m and exceeds 27 hours with progressive renal impairment. The half-life in the elderly was significantly longer (8.8 + or - 0.9 hr) compared with that in the young (5.8 + or - 1.1 hr) (p < 0.01). |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Atenolol therapy has been associated with mild-to-moderate elevations of serum aminotransferase levels in 1% to 2% of patients. These elevations, however, are usually asymptomatic and transient and resolve even with continuation of therapy. A few instances of clinically apparent, acute liver injury attributable to atenolol have been reported. In view of its wide scale use, atenolol induced liver injury is exceedingly rare. The onset of injury has been within 1 to 4 weeks and pattern of liver enzyme elevations has been hepatocellular or mixed. Symptoms of hypersensitivity (rash, fever, eosinophilia) are uncommon as is autoantibody formation. Most cases are self-limiting and resolve rapidly once atenolol is stopped; however, at least one fatal instance has been reported. Likelihood score: D (Possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because of atenolol's relatively extensive excretion into breastmilk and its extensive renal excretion, other agents may be preferred while nursing a newborn or preterm infant or with high maternal dosages. Infants older than 3 months of age appear to be at little risk of adverse effects from atenolol in breastmilk. Timing breastfeeding with respect to the time of the atenolol dose appears to be of little benefit in reducing infant atenolol exposure because the time of the peak is unpredictable. ◉ Effects in Breastfed Infants A study of mothers taking beta-blockers during nursing found a numerically, but not statistically significant increased number of adverse reactions in those taking any beta-blocker. Although the ages of infants were matched to control infants, the ages of the affected infants were not stated. Of 13 mothers taking atenolol, one reported lethargy in her breastfed infant; she was also taking other unspecified drugs for hypertension. Cyanosis, bradycardia and hypothermia occurred in a 5-day-old infant probably because of atenolol in breastmilk. Her mother was taking atenolol 50 mg twice daily. Symptoms continued until day 8 when breastfeeding was discontinued. No difference between resting and crying heart rates were observed in 22 breastfed (extent not stated) infants aged 3 to 4 months whose mothers were taking atenolol in an average oral dosage of 49 mg daily. This finding indicated that the infants were experiencing no beta-adrenergic blockade from atenolol in breastmilk. Other authors have reported 15 infants aged 3 days to 2 weeks exposed to atenolol in breastmilk with no signs of adverse effects. Maternal dosages were 50 or 100 mg daily. ◉ Effects on Lactation and Breastmilk One unusual case of oligomenorrhea, hyperprolactinemia and galactorrhea was reported in a 38-year-old woman who had been taking atenolol for about 18 months. Prolactin values returned to normal within 3 days of discontinuation of atenolol. Galactorrhea slowly lessened and disappeared one month after atenolol discontinuation. ◈ What is atenolol? Atenolol is a medication that has been used to treat high blood pressure, chest pain (angina), and heart rhythm issues (arrythmias). It has also been used to treat, prevent, and improve survival after a heart attack. It belongs to the class of medications called beta-blockers. A brand name for atenolol is Tenormin®.Sometimes when people find out they are pregnant, they think about changing how they take their medication, or stopping their medication altogether. However, it is important to with your healthcare providers before making any changes to how you take this medication. Your healthcare providers can talk with you about the benefits of treating your condition and the risks of untreated illness during pregnancy. ◈ I take atenolol. Can it make it harder for me to get pregnant? It is not known if atenolol can make it harder to get pregnant. ◈ Does taking atenolol increase the chance for miscarriage? Miscarriage is common and can occur in any pregnancy for many different reasons. Studies have not been done to see if atenolol increases the chance for miscarriage. ◈ Does taking atenolol increase the chance of birth defects? Every pregnancy starts out with a 3-5% chance of having a birth defect. This is called the background risk. Studies have not been done to see if atenolol increases the chance for birth defects. ◈ Does taking atenolol in pregnancy increase the chance of other pregnancy-related problems? Atenolol has been linked with reduced growth of the fetus (smaller in size and/or low birth weight). It is not clear if this happens because of the medication, the condition being treated, or other factors. One study did find that atenolol can directly affect blood flow through the placenta, which might be linked with poor growth of the fetus, causing low birth weight (weighing less than 5 pounds, 8 ounces [2500 grams] at birth). ◈ Does taking atenolol in pregnancy affect future behavior or learning for the child? Studies have not been done to see if atenolol can cause behavior or learning issues for the child. ◈ Breastfeeding while taking atenolol: Atenolol passes into breastmilk. There have been reports of babies with slow heart rate, low blood pressure, a bluish color in the skin due to a lack of oxygen in the blood (cyanosis), and low body temperature after being exposed to atenolol through breast milk. If you suspect the baby has any symptoms (slow heart rate, low blood pressure, a bluish color in the skin, lips, or fingernails) contact the child’s healthcare provider.The product label for atenolol recommends people who are breastfeeding not use this medication. But, the benefit of using atenolol may outweigh possible risks. Your healthcare providers can talk with you about using atenolol and what treatment is best for you. Be sure to talk to your healthcare provider about all of your breastfeeding questions. ◈ If a male takes atenolol, could it affect fertility (ability to get partner pregnant) or increase the chance of birth defects? Based on the studies reviewed, it is not known if atenolol could affect male fertility or increase the chance of birth defects above the background risk. In general, exposures that fathers or sperm donors have are unlikely to increase risks to a pregnancy. For more information, please see the MotherToBaby fact sheet on Paternal Exposures at https://mothertobaby.org/fact-sheets/paternal-exposures-pregnancy/. Protein Binding 6-16% bound in plasma. Atenolol binds to two sites on human serum albumin. |
| 参考文献 |
|
| 其他信息 |
Atenolol is a beta-selective (cardioselective) adrenoceptor blocking drug without partial agonist or membrane stabilising activity. Its profile of action most closely resembles that of metoprolol which differs only in that it has some membrane stabilising activity. Atenolol has been well studied and is effective in the treatment of hypertension and in the prophylactic management of angina. Its narrow dose response range obviates the need for highly individualised dose titration. In patients with angina its long duration of beta-blocking activity allows once daily dosage, whereas other beta-blockers, unless in sustained release dosage forms, need to be given in divided doses. Other beta-blockers can be given once daily in hypertension, but at presnt the evidence for effective control with a once daily regimen is more convincing with atenolol. Further studies are need to clarify any important differences in blood pressure control between the various beta-blocking drugs, both in conventional or sustained release dosage forms. As with metoprolol, atenolol is preferable to non-selective beta-blockers in patients with asthma or diabetes mellitus. Atenolol has been well tolerated in most patients, its profile of adverse reactions generally resembling that of other beta-blocking drugs, although its low lipid solubility and limited penetration into the brain results in a lower incidence of central nervous system effects than seen with propranolol. Atenolol is eliminated virtually entirely as unchanged drug in the urine and dosage needs to be reduced in patients with moderate to severely impaired renal function (glomerular filtration rate less than 30 ml/min). There is no need for modification of dosage of atenolol in liver disease.[1]
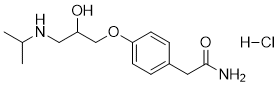
Atenolol can cause developmental toxicity according to state or federal government labeling requirements. Atenolol is an ethanolamine compound having a (4-carbamoylmethylphenoxy)methyl group at the 1-position and an N-isopropyl substituent. It has a role as a beta-adrenergic antagonist, an anti-arrhythmia drug, an antihypertensive agent, a sympatholytic agent, a xenobiotic and an environmental contaminant. It is a member of ethanolamines, a monocarboxylic acid amide and a propanolamine. Atenolol is a cardioselective beta-blocker used in a variety of cardiovascular conditions. Sir James Black, a Scottish pharmacologist, pioneered the use of beta-blockers for the management of angina pectoris in 1958 for which he received the Nobel Prize. Beta-blockers quickly became popular in clinical use and where subsequently investigated for use in myocardial infarction, arrhythmias, and hypertension during the 1960s. Later they continued to be investigated for use in heart failure throughout the 1970-1980s. Atenolol itself was developed early on in this history by Alvogen Malta under the trade name Tenormin and received FDA approval in September, 1981. Despite being one of the most widely prescribed beta blockers, evidence suggests atenolol may not significantly reduce mortality, and only modestly reduce the risk of cardiovascular disease in patients with hypertension. A Cochrane review of patients being treated for primary hypertension shows that atenolol shows a risk ratio of 0.88 for cardiovascular disease risk and a risk ratio of 0.99 for mortality. Similar results have been found in other meta-analyses. A meta-analysis of over 145,000 patients showed the risk of stroke in patients taking atenolol may depend on the age of the patient. The use of atenolol may need to be based on more patient factors than hypertension alone. Atenolol is a beta-Adrenergic Blocker. The mechanism of action of atenolol is as an Adrenergic beta-Antagonist. Atenolol is a cardioselective beta-blocker that is widely used in the treatment of hypertension and angina pectoris. Atenolol has been linked to rare cases of drug induced liver injury, some of which have been fatal. Atenolol is a synthetic isopropylamino-propanol derivative used as an antihypertensive, hypotensive and antiarrhythmic Atenolol acts as a peripheral, cardioselective beta blocker specific for beta-1 adrenergic receptors, without intrinsic sympathomimetic effects. It reduces exercise heart rates and delays atrioventricular conduction, with overall oxygen requirements decreasing. (NCI04) Atenolol is a so-called beta1-selective (or 'cardioselective') drug. That means that it exerts greater blocking activity on myocardial beta1-receptors than on beta2 ones in the lung. The beta2 receptors are responsible to keep the bronchial system open. If these receptors are blocked, bronchospasm with serious lack of oxygen in the body can result. However, due to its cardioselective properties, the risk of bronchospastic reactions if using atenolol is reduced compared to nonselective drugs as propranolol. Nonetheless, this reaction may also be encountered with atenolol, particularly with high doses. Extreme caution should be exerted if atenolol is given to asthma patients, who are particularly at risk; the dose should be as low as possible. If an asthma attack occurs, the inhalation of a beta2-mimetic antiasthmatic, such as hexoprenalin or salbutamol, will usually suppress the symptoms. Atenolol (trade name Tenormin) can be used to treat cardiovascular diseases such as hypertension, coronary heart disease, arrhythmias, and treatment of myocardial infarction after the acute event. Patients with compensated congestive heart failure may be treated with atenolol as a co medication (usually together with an ACE inhibitor, a diuretic and a digitalis-glycoside, if indicated). In patients with congestive heart failure, it reduces the need for and the consumption of oxygen of the heart muscle. It is very important to start with low doses, as atenolol reduces also the muscular power of the heart, which is an undesired effect in congestive heart failure. A cardioselective beta-1 adrenergic blocker possessing properties and potency similar to PROPRANOLOL, but without a negative inotropic effect. See also: Atenolol; Chlorthalidone (component of); Atenolol; scopolamine hydrobromide (component of). Drug Indication **Indicated** for: 1) Management of hypertension alone and in combination with other antihypertensives. 2) Management of angina pectoris associated with coronary atherosclerosis. 3) Management of acute myocardial infarction in hemodynamically stable patients with a heart rate greater than 50 beats per minutes and a systolic blood pressure above 100 mmHg. **Off-label** uses include: 1) Secondary prevention of myocardial infarction. 2) Management of heart failure. 3) Management of atrial fibrillation. 4) Management of supraventricular tachycardia. 5) Management of ventricular arrythmias such as congenital long-QT and arrhythmogenic right ventricular cardiomyopathy. 6) Management of symptomatic thyrotoxicosis in combination with [methimazole]. 7) Prophylaxis of migraine headaches. 8) Management of alcohol withdrawal. FDA Label Mechanism of Action Atenolol is a cardioselective beta-blocker, called such because it selectively binds to the β1-adrenergic receptor as an antagonist up to a reported 26 fold more than β2 receptors. Selective activity at the β1 receptor produces cardioselectivity due to the higher population of this receptor in cardiac tissue. Some binding to β2 and possibly β3 receptors can still occur at therapeutic dosages but the effects mediated by antagonizing these are significantly reduced from those of non-selective agents. β1 and β2 receptors are Gs coupled therefore antagonism of their activation reduces activity of adenylyl cyclase and its downstream signalling via cyclic adenosime monophosphate and protein kinase A (PKA). In cardiomyocytes PKA is thought to mediate activation of L-type calcium channels and ryanodine receptors through their phosphorylation. L-type calcium channels can then provide an initial rise in intracellular calcium and trigger the ryanodine receptors to release calcium stored in the sarcoplasmic reticulum (SR) and increased contractility. PKA also plays a role in the cessation of contraction by phosphorylating phospholamban which in turn increases the affinity of SR Ca2+ ATPase to increase reuptake of calcium into the SR. It also phophorylates troponin I to reduce affinity of the protein for calcium. Both of these events lead to a reduction in contraction which, when coupled with the initial increase in contraction, allows for faster cycling and consequently higher heart rate with increased contractility. L-type calcium channels are also a major contributor to cardiac depolarization and their activation can increase frequency of action potentials and possibly the incidence of ectopic potentials. Similar inihibitory events occur in the bronchial smooth muscle to mediate relaxation including phosphorylation of myosin light-chain kinase, reducing its affinity for calcium. PKA also inhibits the excitatory Gq coupled pathway by phosphorylating the inositol trisphosphate receptor and phospholipase C resulting in inhibition of intracellular calcium release. Antagonism of this activity by beta-blocker agents like atenolol can thus cause increased bronchoconstriction. By inhibiting myocardial beta 1-adrenergic receptors, atenolol produces negative chronotropic and inotropic activity. The negative chronotropic action of atenolol on the sinoatrial node results in a decrease in the rate of sinoatrial node discharge and an increase in recovery time, thereby decreasing resting and exercise stimulated heart rate and reflex orthostatic tachycardia by about 25-35%. High doses of the drug may produce sinus arrest, especially in patients with sinoatrial node disease (eg, sick sinus syndrome). Atenolol also slows conduction in the atrioventricular nose. Although stroke index may be increased moderately by about 10%, atenolol usually reduces cardiac output by about 20% probably secondary to its effect on heart rate. The decrease in myocardial contractability and heart rate, as well as the reduction in blood pressure, produced by atenolol generally lead to a reduction in myocardial oxygen consumption which accounts for the effectiveness of the drug in chronic stable angina pectoris; however, atenolol can increase oxygen requirements by increasing left ventricular fiber length and end-diastolic pressure, particularly in patients with cardiac failure. Atenolol suppresses plasma renin activity and suppresses the renin aldosterone angiotensin system. The toxic actions of beta-blockers appear to be related to properties such as membrane depressant activity and possibly due to actions on beta-adrenoceptors distinct from those in the cardiovascular system. |
| 分子量 |
302.79702
|
|---|---|
| 精确质量 |
302.14
|
| 元素分析 |
C, 55.53; H, 7.66; Cl, 11.71; N, 9.25; O, 15.85
|
| CAS号 |
51706-40-2
|
| 相关CAS号 |
Atenolol-d7;1202864-50-3; 51706-40-2 (HCl); 29122-68-7;93379-54-5 (S isomer); 56715-13-0 (R isomer)
|
| PubChem CID |
119274
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.125g/cm3
|
| 沸点 |
508ºC at 760mmHg
|
| 闪点 |
261.1ºC
|
| LogP |
2.794
|
| tPSA |
85.57
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
20
|
| 分子复杂度/Complexity |
263
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC(C)NCC(COC1=CC=C(C=C1)CC(=O)N)O.Cl
|
| InChi Key |
FFDDLJYKJQGSPW-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C14H22N2O3.ClH/c1-10(2)16-8-12(17)9-19-13-5-3-11(4-6-13)7-14(15)18;/h3-6,10,12,16-17H,7-9H2,1-2H3,(H2,15,18);1H
|
| 化学名 |
2-[4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]acetamide;hydrochloride
|
| 别名 |
Atenolol hydrochloride; 51706-40-2; Atenolol HCl; dl-Atenolol.HCl; 4-[2-HYDROXY-3-[(ISOPROPYL)AMINO]PROPOXY]PHENYLACETAMIDE HYDROCHLORIDE; 6DI0UT7U1Q; 4-(2-Hydroxy-3-((isopropyl)amino)propoxy)phenylacetamide hydrochloride; 2-[4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]acetamide hydrochloride;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3025 mL | 16.5125 mL | 33.0251 mL | |
| 5 mM | 0.6605 mL | 3.3025 mL | 6.6050 mL | |
| 10 mM | 0.3303 mL | 1.6513 mL | 3.3025 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04931108 | RECRUITING | Drug: Nitrendipine/Atenolol Drug: Nitrendipine Drug: Atenolol |
Hypertension | Shanghai Jiao Tong University School of Medicine | 2021-09-27 | Phase 4 |
| NCT00913965 | COMPLETED | Drug: Atenolol Tablets 100 mg (Cord Laboratories) Drug: Atenolol Tablets 100 mg (Stuart Pharmaceutical) |
Hypertension | Sandoz | 1989-07 | Phase 1 |
| NCT01719367 | COMPLETEDWITH RESULTS | Drug: Atenolol | Atrial Fibrillation | Vanderbilt University Medical Center | 2013-01 | Not Applicable |
| NCT01397994 | UNKNOWN STATUS | Drug: Nicorandil Drug: Atenolol |
Chronic Stable Angina | Ferozsons Laboratories Ltd. | 2011-09 | Phase 4 |
| NCT04905277 | ACTIVE, NOT RECRUITING | Drug: Atenolol 50 MG Drug: Placebo |
Healthy | Sundeep Khosla, M.D. | 2021-07-27 | Phase 2 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































