| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| Other Sizes |
|

| 靶点 |
5-HT1A Receptor ( Ki = 4.2 nM ); 5-HT2A Receptor; 5-HT2B Receptor; 5-HT2C Receptor; D2 Receptor; D3 Receptor; D4 Receptor
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
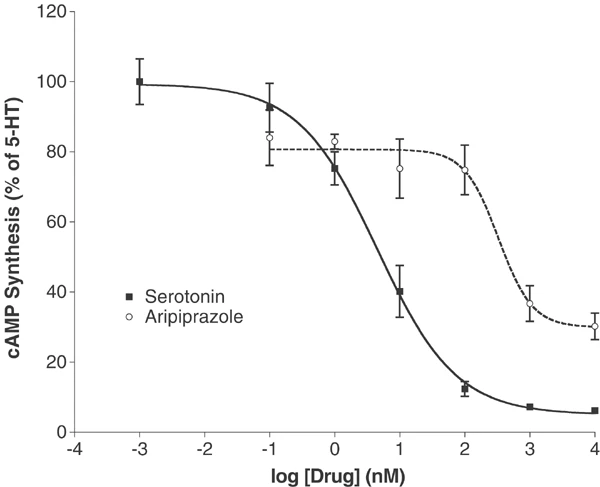
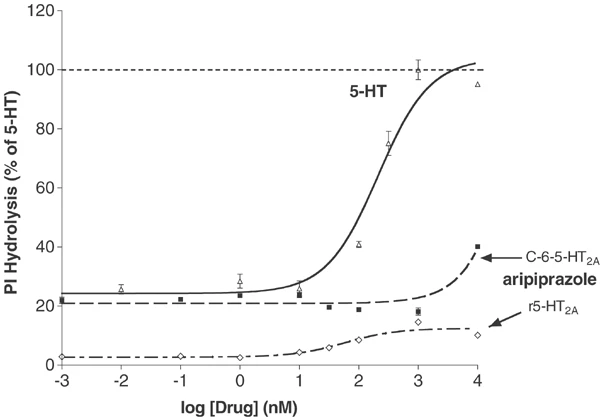
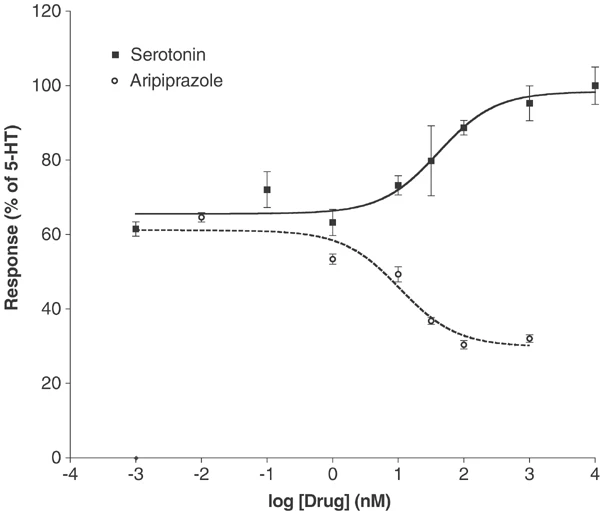
体外活性:阿立哌唑以高亲和力与 G 蛋白偶联和非偶联状态的受体结合。阿立哌唑可有效激活 D2 受体介导的 cAMP 积累抑制作用。阿立哌唑对 h5-HT(2B)-、hD(2L)- 和 hD(3)- 多巴胺受体具有最高的亲和力,但对其他几种 5-HT 受体 (5-HT) 也具有显着的亲和力 (5-30 nM) (1A)、5-HT(2A)、5-HT(7)),以及 α(1A)-肾上腺素能受体和 hH(1)-组胺受体。阿立哌唑对其他 G 蛋白偶联受体的亲和力较低 (30-200 nM),包括 5-HT(1D)、5-HT(2C)、α(1B)-、α(2A)-、α(2B) -、α(2C)-、β(1)-、β(2)-肾上腺素能受体和 H(3)-组胺受体。阿立哌唑是 5-HT(2B) 受体的反向激动剂,并对 5-HT(2A)、5-HT(2C)、D(3) 和 D(4) 受体显示部分激动作用。
|
||
| 体内研究 (In Vivo) |
阿立哌唑可降低未接受药物治疗的大鼠内侧前额叶皮层和纹状体中细胞外 5-HIAA 的浓度,但不会降低慢性阿立哌唑预处理大鼠的浓度。阿立哌唑,0.1 mg/kg 和 0.3 mg/kg,显着增加大鼠海马的多巴胺释放。阿立哌唑,0.3 mg/kg,轻微但显着地增加内侧前额皮质中的多巴胺释放,但不增加伏核中的多巴胺释放。阿立哌唑,3.0 mg/kg 和 10 mg/kg,显着减少伏隔核中的多巴胺释放,但不影响医学前额皮质。阿立哌唑,0.3 mg/kg,可短暂增强氟哌啶醇(0.1 mg/kg)诱导的内侧前额叶皮质中的多巴胺释放,但抑制伏隔核中的多巴胺释放。
|
||
| 酶活实验 |
放射性配体结合试验[2]
大量瞬时和稳定转染的克隆人类cdna,通过国家精神卫生研究所精神活性药物筛选计划(NIMH-PDSP)的资源获得,用于放射配体结合和功能分析,如前面所述(Rothman等人,2000;Tsai et al ., 2000)。表1列出了放射配体结合测定的条件,以及标准化合物的KD值。在初始筛选试验中,以10 μM的浓度对大量gpcr、离子通道和转运体进行了阿立哌唑四次重复的测试。对于>50%抑制的分子靶点,使用至少6个浓度<强>阿立哌唑的浓度来测定Ki;使用GraphPad Prism计算四份Ki值。[125I]DOI竞争试验按照前面的描述进行(Choudhary等,1992),并做了以下改变:将12个阿立哌唑的稀释度,范围为0.01-3000 nM,与[125I]DOI (0.3 nM)在25°C下,以总体积为0.25 ml,结合缓冲液(50 mM Tris缓冲液,pH 7.4, 0.5 mM EDTA, 10 mM MgCl2)中5-20 μg的膜蛋白孵育1小时。用Brandel细胞收割机在聚乙烯亚胺预处理(0.3%)Whatman GF/C过滤器上进行三次冷水洗涤,收获膜。结合滤光片的放射性是用液体闪烁计数来量化的。 阿立哌唑是第一个下一代非典型抗精神病药,其作用机制不同于目前上市的典型和非典型抗精神病药。阿立哌唑分别在多巴胺能低活性和多活性动物模型中表现出激动剂和拮抗剂的特性。本研究检测了阿立哌唑与单一人群D2受体的相互作用,以进一步阐明其药理学特性。在表达重组D2L受体的中国仓鼠卵巢细胞制备的膜中,阿立哌唑对G蛋白偶联和非偶联状态的受体都具有高亲和力。阿立哌唑有效激活D2受体介导的cAMP积累抑制。用烷基化剂n -乙氧羰基-2-乙氧基-1,2-二氢喹啉(EEDQ)灭活部分受体显著降低了阿立哌唑抑制cAMP积累的最大效果。这种效应是在EEDQ浓度不改变多巴胺最大抑制作用的情况下观察到的。与部分激动剂的预期作用一致,增加阿立哌唑浓度阻断多巴胺的作用,其最大阻断作用相当于单独使用阿立哌唑的激动剂作用。阿立哌唑相对于多巴胺的疗效在缺乏多巴胺备用受体的细胞中为25%,在具有受体储备的细胞中为90%。这些结果,连同先前的研究表明部分激动剂对5-羟色胺(5-HT)1A受体的活性和拮抗剂对5-HT2A受体的活性,支持阿立哌唑作为多巴胺- 5-羟色胺系统稳定剂的鉴定。受体活性谱可能是阿立哌唑在动物体内的独特活性及其在人类中的抗精神病活性的基础。[2] |
||
| 细胞实验 |
阿立哌唑对cAMP生成的影响[2]
福斯克林刺激cAMP生成的抑制作用[2] 如先前报道的那样,在稳定的D4和5- ht1a受体表达细胞系中测量了福斯克林刺激的3 ‘,5 ’环腺苷单磷酸(cAMP)产生的抑制作用(Lawler等,1999;Zhang et al ., 1994)。简单地说,在24孔板中培养细胞,在实验之前用含有100 μM IBMX和100 μM forskolin(全部在冰上)的新鲜F12培养基替换生长培养基。在细胞中加入10倍稀释的阿立哌唑 0.1 ~ 10.000 nM,然后在37℃和5% CO2下孵育20 min。通过抽吸和加入0.5 ml的3%三氯乙酸来终止反应。4℃冷冻1 h, 1000 g旋转15 min。cAMP采用竞争性结合测定法进行了少量修改(Nordstedt和Fredholm, 1990)。测定cAMP含量时,将三氯乙酸提取物(40 μl)加入到含有cAMP测定缓冲液(100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM EDTA)的反应管中。[3H]每管加入终浓度为1 nM的cAMP,然后加入cAMP结合蛋白(500 μl cAMP缓冲液中约100 μg牛肾上腺皮质粗提物)。反应管在冰上孵育2小时,然后用Brandel细胞收集机收获到浸泡在水中的Whatman GF/C过滤器上。滤光片干燥,结合放射性通过液体闪烁计数来量化。每个样品中cAMP的浓度从0.1至100 pmol /assay的标准曲线估计。 刺激cAMP产量[2] 使用先前描述的方法在稳定的转染物中研究了5-HT6和5-HT7受体中血清素和阿立哌唑的作用(Max等人,1995;Monsma et al ., 1993;Shen et al ., 1993)。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Tablet: Aripiprazole is well absorbed after administration of the tablet, with peak plasma concentrations occurring within 3 hours to 5 hours; the absolute oral bioavailability of the tablet formulation is 87%. ABILIFY can be administered with or without food. Administration of a 15 mg ABILIFY tablet with a standard high-fat meal did not significantly affect the Cmax or AUC of aripiprazole or its active metabolite, dehydro-aripiprazole, but delayed Tmax by 3 hours for aripiprazole and 12 hours for dehydro-aripiprazole. Oral Solution: Aripiprazole is well absorbed when administered orally as the solution. At equivalent doses, the plasma concentrations of aripiprazole from the solution were higher than that from the tablet formulation. In a relative bioavailability study comparing the pharmacokinetics of 30 mg aripiprazole as the oral solution to 30 mg aripiprazole tablets in healthy subjects, the solution-to-tablet ratios of geometric mean Cmax and AUC values were 122% and 114%, respectively. The single-dose pharmacokinetics of aripiprazole were linear and dose-proportional between the doses of 5 mg to 30 mg. Extended-release injectable suspension, bimonthly injection: Aripiprazole absorption into the systemic circulation is prolonged following gluteal intramuscular injection due to the low solubility of aripiprazole particles. The release profile of aripiprazole from ABILIFY ASIMTUFII results in sustained plasma concentrations over 2 months following gluteal injection(s). Following multiple doses, the median peak:trough ratio for aripiprazole following an ABILIFY ASIMTUFII dose is 1.3, resulting in a flat plasma concentration profile with Tmax ranging between 1 to 49 days following multiple gluteal administrations of 960 mg. Following a single oral dose of [14C]-labeled aripiprazole, approximately 25% and 55% of the administered radioactivity was recovered in the urine and feces, respectively. Less than 1% of unchanged aripiprazole was excreted in the urine and approximately 18% of the oral dose was recovered unchanged in the feces. The steady-state volume of distribution of aripiprazole following intravenous administration is high (404 L or 4.9 L/kg), indicating extensive extravascular distribution. The clearance of aripiprazole was estimated to be 0.8mL/min/kg. Other studies have also reported a clearance rate of 3297±1042mL/hr. Oral availability 87%. Aripiprazole is well absorbed and can be administered with or without food. Administration with a high fat meal did not affect the Cmax or AUC, but delayed Tmax by 3 hours for aripiprazole, and 12 hours for dehydro-aripiprazole. Time to peak concentration: Peak plasma concentrations: within 3 to 5 hours. The steady-state volume of distribution of aripiprazole following intravenous administration is high (404 L or 4.9 L/kg), indicating extensive extravascular distribution. At therapeutic concentrations, aripiprazole and its major metabolite are greater than 99% bound to serum proteins, primarily to albumin. There was dose-dependent D2-receptor occupancy indicating brain penetration of aripiprazole in healthy human volunteers administered 0.5 to 30 mg per day. For more Absorption, Distribution and Excretion (Complete) data for ARIPIPRAZOLE (8 total), please visit the HSDB record page. Metabolism / Metabolites Aripiprazole is metabolized primarily by three biotransformation pathways: dehydrogenation, hydroxylation, and N-dealkylation. Based on in vitro studies, CYP3A4 and CYP2D6 enzymes are responsible for the dehydrogenation and hydroxylation of aripiprazole, and N-dealkylation is catalyzed by CYP3A4. Aripiprazole is the predominant drug moiety in systemic circulation. At steady-state, dehydro-aripiprazole, the active metabolite, represents about 40% of aripiprazole AUC in plasma. Aripiprazole is extensively metabolized in the liver principally via dehydrogenation, hydroxylations, and N-dealkylation by the cytochrome P-450 (CYP) 2D6 and 3A4 isoenzymes. The major active metabolite of aripiprazole, dehydro-aripiprazole, exhibits affinity for D2 receptors similar to that of the parent compound and represents approximately 40% of the aripiprazole area under the concentration-time curve (AUC) in plasma. Steady-state plasma concentrations of both aripiprazole and dehydro-aripiprazole are achieved within 14 days. ABILIFY activity is presumably primarily due to the parent drug, aripiprazole, and to a lesser extent, to its major metabolite, dehydro-aripiprazole, which has been shown to have affinities for D2 receptors similar to the parent drug and represents 40% of the parent drug exposure in plasma. Aripiprazole has known human metabolites that include dehydro-aripiprazole, 4-[(2-oxo-3,4-dihydro-1H-quinolin-7-yl)oxy]butanal, 4-Hydroxyaripiprazole, and 2,3-dichlorophenylpiperazine. Aripiprazole is metabolized primarily by three biotransformation pathways: dehydrogenation, hydroxylation,and N-dealkylation.Based on in vitro studies,CYP3A4 and CYP2D6 enzymes are responsible for dehydrogenation and hydroxylation of aripiprazole, and N-dealkylation is catalyzed by CYP3A4.Aripiprazole is the predominant drug moiety in the systemic circulation. At steady-state, dehydro-aripiprazole, the active metabolite, represents about 40% of aripiprazole AUC in plasma (RxList, A308). Route of Elimination: Less than 1% of unchanged aripiprazole was excreted in the urine and approximately 18% of the oral dose was recovered unchanged in the feces. Half Life: 75-146 hours Biological Half-Life The mean elimination half-lives are about 75 hours and 94 hours for aripiprazole and dehydro-aripiprazole, respectively. For populations that are poor CYP2D6 metabolizers, the half-life of aripiprazole is 146 hours and these patients should be treated with half the normal dose. Other studies have reported a half-life of 61.03±19.59 hours for aripiprazole and 279±299 hours for the active metabolite. The mean elimination half-lives are about 75 hours and 94 hours for aripiprazole and dehydro-aripiprazole, respectively. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Aripiprazole is used IM for the acute management of agitation associated with schizophrenia or bipolar disorder, mixed or manic, in adults for whom treatment with aripiprazole is appropriate and who require an IM antipsychotic agent for rapid control of behaviors that interfere with diagnosis and care (e.g., threatening behaviors, escalating or urgently distressing behavior, self-exhausting behavior). Aripiprazole is used orally for the acute treatment of irritability associated with autistic disorder. Aripiprazole is used orally as an adjunct to antidepressants for the acute treatment of major depressive disorder in adults. Aripiprazole is used orally as monotherapy or as an adjunct to either lithium or valproate for the acute treatment of manic or mixed episodes associated with bipolar I disorder with or without psychotic features in adults and pediatric patients 10-17 years of age. The drug also is used orally as monotherapy or as adjunctive therapy with lithium or valproate for the maintenance treatment of bipolar I disorder in adults and pediatric patients 10-17 years of age. Aripiprazole is used orally for the acute and maintenance treatment of schizophrenia in adults and adolescents 13-17 years of age. HUMAN EXPOSURE AND TOXICITY: A potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) has been reported in association with administration of antipsychotic drugs, including aripiprazole. Two possible cases of NMS occurred during aripiprazole treatment in the premarketing worldwide clinical database. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmia). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. ANIMAL STUDIES: In female mice, the incidences of pituitary gland adenomas and mammary gland adenocarcinomas and adenoacanthomas were increased at dietary dose of 3 to 30 mg/kg/day. Female rats were treated with oral doses of 2, 6, and 20 mg/kg/day of aripiprazole from 2 weeks prior to mating through day 7 of gestation. Estrus cycle irregularities and increased corpora lutea were seen at all doses, but no impairment of fertility was seen. Increased pre-implantation loss was seen at 6 and 20 mg/kg, and decreased fetal weight was seen at 20 mg/kg. Pregnant rabbits were treated with oral doses of 10, 30, and 100 mg/kg/day of aripiprazole during the period of organogenesis. Decreased maternal food consumption and increased abortions were seen at 100 mg/kg. Treatment caused increased fetal mortality (100 mg/kg), decreased fetal weight (30 and 100 mg/kg), increased incidence of a skeletal abnormality (fused sternebrae at 30 and 100 mg/kg) and minor skeletal variations (100 mg/kg). Aripiprazole and a metabolite (2,3-DCPP) were clastogenic in the in vitro chromosomal aberration assay in CHL cells with and without metabolic activation. The metabolite, 2,3-DCPP, produced increase in numerical aberrations in the in vitro assay in CHL cells in the absence of metabolic activation. A positive response was obtained in the in vivo micronucleus assay in mice, however,the response was shown to be due to a mechanism not considered relevant to humans. Aripiprazole's antipsychotic activity is likely due to a combination of antagonism at D2 receptors in the mesolimbic pathway and 5HT2A receptors in the frontal cortex. Antagonism at D2 receptors relieves positive symptoms while antagonism at 5HT2A receptors relieves negative symptoms of schizophrenia. Aripiprazole exhibits high affinity for dopamine D2 and D3, serotonin 5-HT1A and 5- HT2A receptors, moderate affinity for dopamine D4, serotonin 5-HT2C and 5-HT7, alpha1-adrenergic and histamine H1 receptors and moderate affinity for the serotonin reuptake pump. Aripiprazole has no appreciable affinity for cholinergic muscarinic receptors. Aripiprazole functions as a partial agonist at the dopamine D2 and the serotonin 5-HT1A receptors, and as an antagonist at serotonin 5-HT2A receptor. Interactions Substrates of Hepatic Microsomal Enzymes: Substrates of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4; pharmacokinetic interaction unlikely. Hypotensive Agents: Potential pharmacologic interaction (additive hypotensive effects) Famotidine: Coadministration of aripiprazole (given in a single dose of 15 mg) with a 40-mg single dose of the H2 antagonist famotidine, a potent gastric acid blocker, decreased the solubility of aripiprazole and, hence, its rate of absorption, reducing by 37% and 21% the Cmax of aripiprazole and dehydro-aripiprazole, respectively, and by 13% and 15%, respectively, the extent of absorption (AUC). No dosage adjustment of aripiprazole is required when administered concomitantly with famotidine. Valproate: When valproate (500-1500 mg/day) and aripiprazole (30 mg/day) were coadministered at steady state, the C max and AUC of aripiprazole were decreased by 25%. No dosage adjustment of aripiprazole is required when administered concomitantly with valproate. For more Interactions (Complete) data for ARIPIPRAZOLE (12 total), please visit the HSDB record page. |
||
| 参考文献 | |||
| 其他信息 |
Therapeutic Uses
Antipsychotic Agents Aripiprazole is used IM for the acute management of agitation associated with schizophrenia or bipolar disorder, mixed or manic, in adults for whom treatment with aripiprazole is appropriate and who require an IM antipsychotic agent for rapid control of behaviors that interfere with diagnosis and care (e.g., threatening behaviors, escalating or urgently distressing behavior, self-exhausting behavior). Aripiprazole is used orally for the acute treatment of irritability associated with autistic disorder. Aripiprazole is used orally as an adjunct to antidepressants for the acute treatment of major depressive disorder in adults. For more Therapeutic Uses (Complete) data for ARIPIPRAZOLE (6 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS. Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of seventeen placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (eg, heart failure, sudden death) or infectious (eg, pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear. ABILIFY (aripiprazole) is not approved for the treatment of patients with dementia-related psychosis. /Included in label/ /BOXED WARNING/ WARNING: INCREASED SUICIDALTHOUGHTS AND BEHAVIORS. Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Anyone considering the use of adjunctive ABILIFY or any other antidepressant in a child, adolescent, or young adult must balance this risk with the clinical need. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction in risk with antidepressants compared to placebo in adults aged 65 and older. Depression and certain other psychiatric disorders are themselves associated with increases in the risk of suicide. Patients of all ages who are started on antidepressant therapy should be monitored appropriately and observed closely for clinical worsening, suicidality, or unusual changes in behavior. Families and caregivers should be advised of the need for close observation and communication with the prescriber. ABILIFY is not approved for use in pediatric patients with depression. /Included in label/ Contraindications: Known hypersensitivity reaction to aripiprazole or any ingredient in the formulation; such reactions have ranged from pruritus/urticaria to anaphylaxis. Safety and effectiveness in pediatric patients with bipolar mania were established in a 4-week, placebo-controlled clinical trial in 197 pediatric patients aged 10 to 17 years. The incidence of discontinuation due to adverse reactions between aripiprazole-treated and placebo-treated pediatric patients (10 to 17 years) was 7% and 2%, respectively. Commonly observed adverse reactions associated with the use of aripiprazole in pediatric patients with bipolar mania (incidence of 5% or greater and aripiprazole incidence at least twice that for placebo) somnolence, extrapyramidal disorder, fatigue, nausea, akathisia, blurred vision, salivary hypersecretion, and dizziness. Although maintenance efficacy in pediatric patients has not been systematically evaluated, maintenance efficacy can be extrapolated from adult data along with comparisons of aripiprazole pharmacokinetic parameters in adult and pediatric patients. For more Drug Warnings (Complete) data for ARIPIPRAZOLE (27 total), please visit the HSDB record page. Pharmacodynamics Aripiprazole exhibits high affinity for dopamine D2 and D3, serotonin 5-HT1a and 5-HT2a receptors (Ki values of 0.34 nM, 0.8 nM, 1.7 nM, and 3.4 nM, respectively), moderate affinity for dopamine D4, serotonin 5-HT2c and 5-HT7, alpha1-adrenergic and histamine H1 receptors (Ki values of 44 nM, 15 nM, 39 nM, 57 nM, and 61 nM, respectively), and moderate affinity for the serotonin reuptake site (Ki=98 nM). Aripiprazole has no appreciable affinity for cholinergic muscarinic receptors (IC50>1000 nM). |
| 分子式 |
C23H27CL2N3O2
|
|
|---|---|---|
| 分子量 |
448.39
|
|
| 精确质量 |
447.148
|
|
| 元素分析 |
C, 61.61; H, 6.07; Cl, 15.81; N, 9.37; O, 7.14
|
|
| CAS号 |
129722-12-9
|
|
| 相关CAS号 |
Aripiprazole-d8; 1089115-06-9; Aripiprazole (1,1,2,2,3,3,4,4-d8); 1089115-04-7; Aripiprazole monohydrate; 851220-85-4; 1259305-26-4 (cavoxil)
|
|
| PubChem CID |
60795
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
646.2±55.0 °C at 760 mmHg
|
|
| 熔点 |
139°C
|
|
| 闪点 |
344.6±31.5 °C
|
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
|
| 折射率 |
1.593
|
|
| LogP |
5.59
|
|
| tPSA |
44.81
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
30
|
|
| 分子复杂度/Complexity |
559
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O=C1NC2=C(C=CC(OCCCCN3CCN(C4=CC=CC(Cl)=C4Cl)CC3)=C2)CC1
|
|
| InChi Key |
CEUORZQYGODEFX-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C23H27Cl2N3O2/c24-19-4-3-5-21(23(19)25)28-13-11-27(12-14-28)10-1-2-15-30-18-8-6-17-7-9-22(29)26-20(17)16-18/h3-6,8,16H,1-2,7,9-15H2,(H,26,29)
|
|
| 化学名 |
7-[4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butoxy]-3,4-dihydro-1H-quinolin-2-one
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.58 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.58 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 View More
配方 3 中的溶解度: 2.5 mg/mL (5.58 mM) in 10% DMF 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 配方 4 中的溶解度: 2.5 mg/mL (5.58 mM) in 10% DMF 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2302 mL | 11.1510 mL | 22.3020 mL | |
| 5 mM | 0.4460 mL | 2.2302 mL | 4.4604 mL | |
| 10 mM | 0.2230 mL | 1.1151 mL | 2.2302 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Maintenance Electroconvulsive Therapy (ECT) Versus Aripiprazole in Clozapine-resistant Schizophrenia
CTID: NCT06501339
Phase: Phase 4 Status: Not yet recruiting
Date: 2024-07-16
|
|
|
 Methamnetamine hydrochloride
Methamnetamine hydrochloride
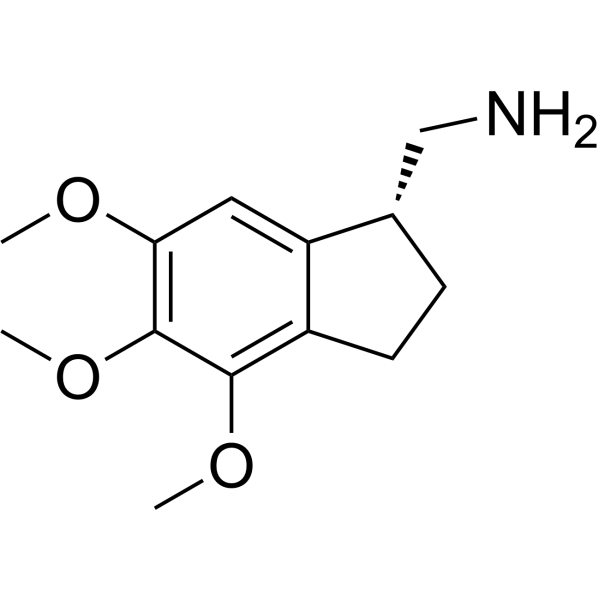 Jimscaline
Jimscaline
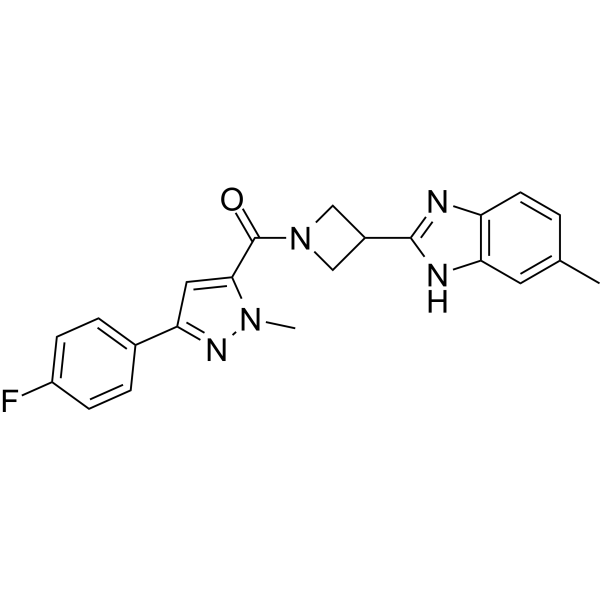 VU0530244
VU0530244
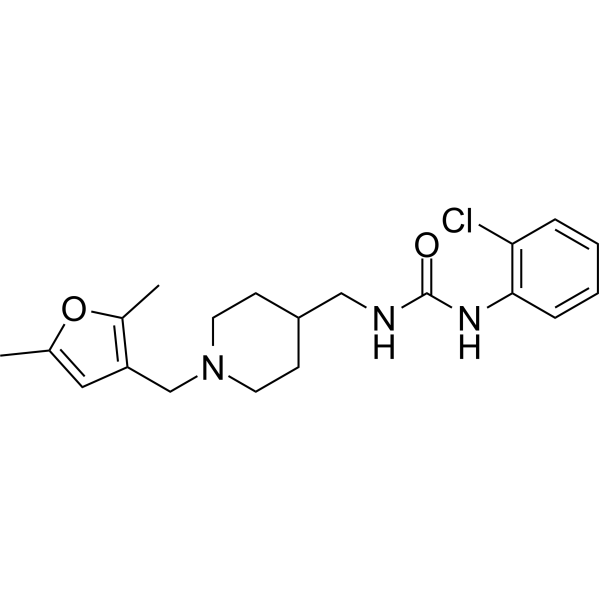 VU0631019
VU0631019
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA