| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
EGFRL858R (IC50 = 0.4 nM); EGFR (wt) (IC50 = 0.5 nM); ErbB4 (IC50 = 1 nM); EGFRL858R/T790M (IC50 = 10 nM); HER2 (IC50 = 14 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:BIBW2992 对野生型和突变型 EGFR 和 HER2 均显示出有效的活性。它针对 L858R EGFR 的效力与吉非替尼相似,但针对吉非替尼耐药的 L858R-T790M EGFR 双突变体的活性高出约 100 倍。 BIBW2992 对体内 EGFR 和 HER2 磷酸化表现出有效的作用。在所有测试的细胞类型中,例如表达 wt EGFR 的人表皮样癌细胞系 A431、转染 wt HER2 的鼠 NIH-3T3 细胞以及乳腺癌细胞系 BT,其与参考化合物(例如 Lapatinib 等)相比均具有优势-474和胃癌细胞系NCI-N87,表达内源性HER2。激酶测定:将人 EGFR 的野生型酪氨酸激酶结构域以及 EGFR L858R/T790M 双突变体的酪氨酸激酶结构域与谷胱甘肽-S-转移酶 (GST) 融合并提取。然后在抑制剂 BIBW2992(在 50% DMSO 中连续稀释)存在的情况下测定酶活性。使用随机聚合物 pEY (4:1) 作为底物,并添加生物素化 pEY (bio-pEY) 作为示踪底物。使用杆状病毒系统克隆 HER2 的激酶结构域,并以与 EGFR 激酶类似的方式进行提取。补充信息中提供了 EGFR、HER2、SRC、BIRK 和 VEGFR2 激酶活性测定的详细信息。细胞测定:将 1 × 104 个 NSCLC 细胞转移至 96 孔板的每个孔中,并在无血清培养基中培养过夜,用于 EGFR 磷酸化测定。第二天添加 BIBW2992 后,将板在 37°C 下孵育 1 小时。 EGF 刺激使用 100 ng/mL 在室温下进行 10 分钟。用冰冷的 PBS 洗涤细胞,每孔用 120 μL HEPEX 缓冲液提取,并在室温下摇动 1 小时。每孔全部 2 × 104 个细胞用于 HER2 磷酸化测定。链霉亲和素预包被板用封闭缓冲液中 1:100 稀释的抗 EGFR-生物素和 c-erb2/HER2 癌蛋白 Ab-5(克隆 N24)-生物素包被。然后将细胞提取物转移至抗体包被的孔中并在室温下孵育1小时。消光在 450 nm 处测量。
|
| 体内研究 (In Vivo) |
每日口服 20 mg/kg 的 BIBW2992 持续 25 天可导致肿瘤显着消退,累积治疗/对照肿瘤体积比(T/C 比)为 2%。通过组织切片的免疫组织化学染色证实 EGFR 和 AKT 磷酸化的减少。因此,与拉帕替尼和来那替尼一样,BIBW2992是下一代酪氨酸激酶抑制剂(TKI),可不可逆地抑制人表皮生长因子受体2(Her2)和表皮生长因子受体(EGFR)激酶。 BIBW2992 不仅能有效对抗厄洛替尼或吉非替尼等第一代 TKI 靶向的 EGFR 突变,还能对抗那些对这些标准疗法不敏感的患者。
|
| 酶活实验 |
将人 EGFR 野生型和 EGFR L858R/T790M 双突变酪氨酸激酶结构域与 GST 融合并提取。接下来,在使用和不使用抑制剂 BIBW2992 的情况下测量酶活性,BIBW2992 在 50% DMSO 中连续稀释。添加生物素化 pEY (bio-pEY) 作为示踪底物,并使用随机聚合物 pEY (4:1) 作为底物。利用杆状病毒系统,以类似于 EGFR 激酶的方式克隆和提取 HER2 激酶结构域。补充信息包含有关 EGFR、HER2、SRC、BIRK 和 VEGFR2 激酶活性检测的具体信息。
|
| 细胞实验 |
对于 EGFR 磷酸化测试,将 1 × 104 NSCLC 细胞接种到 96 孔板的每个孔中,并在无血清培养基中生长整夜。第二天,添加 BIBW2992 后,将板在 37°C 下孵育一小时。 EGF 刺激在室温下使用 100 ng/mL 进行 10 分钟。在室温下振荡一小时并使用每孔 120 μL HEPEX 缓冲液提取后,用冰冷的 PBS 清洗细胞。 HER2 磷酸化检测每孔总共使用 2 × 104 细胞。 c-erb2/HER2 癌蛋白 Ab-5(克隆 N24)-生物素和抗 EGFR-生物素以 1:100 稀释在封闭缓冲液中包被在链霉亲和素预包被板上。一旦进入抗体包被的孔中,细胞提取物就可以在室温下静置一小时。消光测量发生在 450 nm 处。
|
| 动物实验 |
Athymic NMRI-nu/nu female mice[1]
20 mg/kg Oral administration Four bitransgenic mice on continuous doxycycline diets for more than 6 weeks were subjected to MRI (Figure 4) to document the lung tumor burden. Afatinib (BIBW2992) formulated in 0.5% methocellulose-0.4% polysorbate-80 (Tween 80) was administered orally by gavage at 20 mg/kg once daily dosing schedule. Rapamycin was dissolved in 100% ethanol, freshly diluted in 5% PEG400 and 5% Tween 80 before treatment and administered by intraperitoneal injection at 2 mg/kg daily dosage. Mice were monitored by MRI every 1 or 2 weeks to determine reduction in tumor volume and killed for further histological and biochemical studies after drug treatment. For immunohistochemistry staining, three tumor-bearing mice in each group were treated three times with either Afatinib (BIBW2992) (20 mg/kg) alone or Afatinib (BIBW2992) (20 mg/kg) and rapamycin 2 mg/kg at 24 h intervals and killed 1 h after the last drug delivery. All the mice were kept on the doxycycline diet throughout the experiments. Littermates were used as controls.[1] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Following oral administration, time to peak plasma concentration (Tmax) is 2 to 5 hours. Maximum concentration (Cmax) and area under the concentration-time curve from time zero to infinity (AUC0-∞) values increased slightly more than dose proportional in the range of 20 to 50 mg. The geometric mean relative bioavailability of 20 mg tablets was 92% as compared to an oral solution. Additionally, systemic exposure to afatinib is decreased by 50% (Cmax) and 39% (AUC0-∞), when administered with a high-fat meal compared to administration in the fasted state. Based on population pharmacokinetic data derived from clinical trials in various tumor types, an average decrease of 26% in AUCss was observed when food was consumed within 3 hours before or 1 hour after taking afatinib. In humans, excretion of afatinib is primarily via the feces. Following administration of an oral solution of 15 mg afatinib, 85.4% of the dose was recovered in the feces and 4.3% in urine. The parent compound afatinib accounted for 88% of the recovered dose. The volume of distribution of afatinib recorded in healthy male volunteers is documented as 4500 L. Such a high volume of distribution in plasma suggests a potentially high tissue distribution. The apparent total body clearance of afatinib as recorded in healthy male volunteers is documented as being a high geometric mean of 1530 mL/min. Metabolism / Metabolites Enzyme-catalyzed metabolic reactions play a negligible role for afatinib in vivo. Covalent adducts to proteins were the major circulating metabolites of afatinib. Biological Half-Life Afatinib is eliminated with an effective half-life of approximately 37 hours. Thus, steady-state plasma concentrations of afatinib were achieved within 8 days of multiple dosing of afatinib resulting in an accumulation of 2.77-fold (AUC0-∞) and 2.11-fold (Cmax). In patients treated with afatinib for more than 6 months, a terminal half-life of 344 h was estimated. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Elevations in serum aminotransferase levels are common during afatinib therapy occurring in 20% to 50% of patients, but rising above 5 times the upper limit of the normal range in only 1% to 2%. Hepatic failure is said to have occurred in 0.2% of patients and to have resulted in several fatalities. Hepatotoxicity appears to be a class effect among protein kinase inhibitors of EGFR2, although liver injury appears to be more frequent and more severe with gefitinib than with afatinib and erlotinib. Specific details of the liver injury associated with afatinib such as latency, serum enzyme pattern, clinical features and course, have not been published. Other EGFR inhibitors, such as erlotinib and gefitinib typically cause liver injury arising within days or weeks of starting therapy and presenting abruptly with hepatocellular enzyme elevations and a moderate-to-severe course. Immunoallergic and autoimmune features are not common. The rate of clinically significant liver injury and hepatic failure is increased in patients with preexisting cirrhosis or hepatic impairment due to liver tumor burden. Likelihood score: D (possible cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of afatinib during breastfeeding. Because afatinib is about 95% bound to plasma proteins, the amount in milk is likely to be low. However, its half-life is about 37 hours and it might accumulate in the infant. the manufacturer recommends that breastfeeding be discontinued during afatinib therapy and for 2 weeks after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding In vitro binding of afatinib to human plasma proteins is approximately 95%. Afatinib binds to proteins both non-covalently (traditional protein binding) and covalently. |
| 参考文献 | |
| 其他信息 |
Pharmacodynamics
Aberrant ErbB signaling triggered by receptor mutations, and/or amplification, and/or receptor ligand overexpression contributes to the malignant phenotype. Mutation in EGFR defines a distinct molecular subtype of lung cancer. In non-clinical disease models with ErbB pathway deregulation, afatinib as a single agent effectively blocks ErbB receptor signaling resulting in tumor growth inhibition or tumor regression. NSCLC tumors with common activating EGFR mutations (Del 19, L858R) and several less common EGFR mutations in exon 18 (G719X) and exon 21 (L861Q) are particularly sensitive to afatinib treatment in non-clinical and clinical settings. Limited non-clinical and/or clinical activity was observed in NSCLC tumors with insertion mutations in exon 20. The acquisition of a secondary T790M mutation is a major mechanism of acquired resistance to afatinib and gene dosage of the T790M-containing allele correlates with the degree of resistance in vitro. The T790M mutation is found in approximately 50% of patients' tumors upon disease progression on afatinib, for which T790M targeted EGFR TKIs may be considered as a next line treatment option. Other potential mechanisms of resistance to afatinib have been suggested preclinically and MET gene amplification has been observed clinically. At the same time, the effect of multiple doses of afatinib (50 mg once daily) on cardiac electrophysiology and the QTc interval was evaluated in an open-label, single-arm study in patients with relapsed or refractory solid tumors. Ultimately, no large changes in the mean QTc interval (i.e., >20 ms) were detected in the study. |
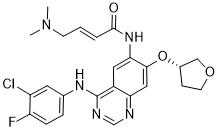
| 分子式 |
C24H25CLFN5O3
|
|---|---|
| 分子量 |
485.94
|
| 精确质量 |
485.162
|
| 元素分析 |
C, 59.32; H, 5.19; Cl, 7.30; F, 3.91; N, 14.41; O, 9.88
|
| CAS号 |
850140-72-6
|
| 相关CAS号 |
Afatinib dimaleate;850140-73-7;Afatinib-d6;1313874-96-2;Afatinib oxalate;1398312-64-5;(R)-Afatinib;439081-17-1;Afatinib-d4
|
| PubChem CID |
10184653
|
| 外观&性状 |
White to light yellow solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
676.9±55.0 °C at 760 mmHg
|
| 熔点 |
100 - 102 °C
|
| 闪点 |
363.2±31.5 °C
|
| 蒸汽压 |
0.0±2.1 mmHg at 25°C
|
| 折射率 |
1.668
|
| LogP |
3.59
|
| tPSA |
88.61
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
34
|
| 分子复杂度/Complexity |
702
|
| 定义原子立体中心数目 |
1
|
| SMILES |
N(C1C=CC(F)=C(Cl)C=1)C1=NC=NC2=CC(=C(C=C12)NC(=O)/C=C/CN(C)C)O[C@@H]1COCC1
|
| InChi Key |
ULXXDDBFHOBEHA-CWDCEQMOSA-N
|
| InChi Code |
InChI=1S/C24H25ClFN5O3/c1-31(2)8-3-4-23(32)30-21-11-17-20(12-22(21)34-16-7-9-33-13-16)27-14-28-24(17)29-15-5-6-19(26)18(25)10-15/h3-6,10-12,14,16H,7-9,13H2,1-2H3,(H,30,32)(H,27,28,29)/b4-3+/t16-/m0/s1
|
| 化学名 |
(E)-N-[4-(3-chloro-4-fluoroanilino)-7-[(3S)-oxolan-3-yl]oxyquinazolin-6-yl]-4-(dimethylamino)but-2-enamide
|
| 别名 |
BIBW2992; Afatinib free base; BIBW 2992; BIBW 2992; Afatinib; trade name: Gilotrif, Tomtovok and Tovok
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.14 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.14 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.14 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2% DMSO+30% PEG 300+5% Tween 80+ddH2O: 10 mg/mL 配方 5 中的溶解度: 5 mg/mL (10.29 mM) in 0.5% Methylcellulose/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0579 mL | 10.2893 mL | 20.5787 mL | |
| 5 mM | 0.4116 mL | 2.0579 mL | 4.1157 mL | |
| 10 mM | 0.2058 mL | 1.0289 mL | 2.0579 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05154396 | Active Recruiting |
Drug: Afatinib | Chordoma | Leiden University Medical Center | June 21, 2018 | Phase 2 |
| NCT03827070 | Active Recruiting |
Drug: Afatinib Drug: Talcum powder |
Non Small Cell Lung Cancer | Center Trials & Treatment Europe | March 5, 2019 | Phase 1 |
| NCT04413201 | Active Recruiting |
Drug: Afatinib Drug: Osimertinib |
Non-squamous NSCLC | Michael Hopp | September 11, 2020 | Phase 4 |
| NCT05267288 | Recruiting | Drug: afatinib | PFS | Qingdao Central Hospital | February 1, 2021 | Phase 2 |
| NCT05519917 | Not yet recruiting | Drug: Afatinib | Chordoma of Clivus | Huashan Hospital | October 1, 2022 | Phase 2 |
|
") |
Afatinib covalently binds to cysteine number 797 of the epidermal growth factor receptor (EGFR) via a Michael addition (IC50 = 0.5 nM).Schubert-Zsilavecz, M, Wurglics, M,Neue Arzneimittel Frühjahr 2013.(in German) |
 PD153035 HCl (SU5271; ZM252868)
PD153035 HCl (SU5271; ZM252868)
 RG 13022
RG 13022
 AV-412
AV-412
 O-去甲基埃罗替尼盐酸盐
O-去甲基埃罗替尼盐酸盐
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
")
")
