| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|
| 体外研究 (In Vitro) |
乙酰紫草素 (1.6-100 μM) 的 IC50 为 40 μM,可抑制 KB-R5 口腔癌细胞的生长 [6]。在 KB-R5 细胞中,乙酰紫草素(20-80 μM;24 小时)除了阻断 mTOR/PI3K/AKT 信号通路外,还可引起细胞凋亡和(20-80 μM)自噬 [6]。通过上调血红素加氧酶-1,乙酰紫草素(1-10 μM;12 h)可以防止 H2O2(500 μM;4 h)诱导的神经母细胞瘤 SH-SY5Y 和 pc12 细胞凋亡 [8]。在吸附/侵袭阶段观察到乙酰紫草素(0.01-5 μM/L;2 h)的抗柯萨奇病毒 A16 (CVA16) 作用,EC50 为 0.04 μmol/L。然而,它在病毒感染前、复制或释放阶段无效[2]。通过 PLC-β3/PKCδ 介导,乙酰紫草素(0.01-1 μM;30 分钟)可增加骨骼肌细胞 L6 的葡萄糖吸收,从而降低血糖水平 [3]。在 HK2 细胞中,乙酰紫草素 (1–5 μg/mL); 48 h) 可以预防 TGF-β1 (5 ng/mL) 引起的肾纤维化 [5]。
|
|---|---|
| 体内研究 (In Vivo) |
在 BALB/c 裸鼠中,乙酰紫草素(50 mg/kg;腹腔注射;每周 3 次,持续六周)显着且浓度依赖性地减少肿瘤的形成 [6]。乙酰紫草素(270–1080 mg/kg;灌胃;每天一次,持续 30 天)可减少 d-半乳糖(150 mg/kg)引起的海马衰老和认知障碍[1]。在小鼠中,乙酰紫草素(2 mg/kg;肌肉注射;单剂量)可抑制柯萨奇病毒 16 (CVA16) 的复制 [2]。乙酰紫草素(100 mg/kg;灌胃;每日一次,持续八周)可通过抑制 TGF-β1/Smad 通路,有效抑制肾纤维化,减少肾功能损伤 [5]。当四氧嘧啶(180 mg/kg;腹腔注射;单剂量)给予糖尿病小鼠时,乙酰紫草素(10 mk/kg;腹腔注射;每天一次,连续三天)会降低血糖水平[3]。在肥胖 C57BL/6J 小鼠中,乙酰紫草素(540 mg/kg;口服;每天一次,持续八周)通过控制脂肪代谢和肝脏炎症来减少肝脂肪的形成,改善肥胖和非酒精性脂肪肝 (NAFLD) [4 ]。乙酰紫草素(120-1080 mg/kg;灌胃;)可通过影响促性腺激素(GTH)分泌并降低血清黄体生成素(LH)和卵泡刺激素(FSH)水平来抑制大鼠妊娠能力。低剂量(120 mg/kg和360 mg/kg)对Sprague-Dawley大鼠无影响,高剂量(1080 mg/kg)对妊娠无影响[7]。
|
| 细胞实验 |
细胞凋亡分析[6]
细胞类型: KB-R5(口腔癌细胞系) 测试浓度: 20 μM、40 μM、80 μM 孵化时间:24小时 实验结果:改变了细胞核的形态。凋亡率增加。 蛋白质印迹分析[6] 细胞类型: KB-R5(口腔癌细胞系) 测试浓度: 20 μM, 40 μM、80 μM 孵育时间: 实验结果: 增加 Beclin-1 和 LC3-II 的表达,抑制 p62 的表达。然而,对LC3-I和Vps34的表达没有影响。以浓度依赖性方式减少 p-mTOR、p-PI3K 和 p-AKT 的表达。 细胞活力测定[2] 细胞类型: CVA16 诱导的人横纹肌肉瘤 (RD) 细胞 测试浓度: 0.01-5 μM/L(与 CVA16 菌株 TA271 1:1 混合) 孵育时间: 2 小时 实验结果:降低 CVA16 诱导的细胞病变效应和抑制率浓度为 0.08 μmol/L 时为 80%。 蛋白质印迹分析 [3] 细胞类型: L6(大鼠骨骼肌细胞) 测试浓度: 0.01 μM、0.1 μM , 1 μM 孵育时间: 2 小时 实验结果: 磷酸盐显着上调 |
| 动物实验 |
Animal/Disease Models: D-galactose (D-gal)-induced sub-acuteaging mouse model of Alzheimer's disease (AD)[1]
Doses: 270 mg/kg, 540 mg/kg, 1080 mg/kg Route of Administration: intragastric (po) administration (ig) ; one time/day for 30 days. After D-gal treatment (150 mg/kg; subcutaneous (sc) injection; one time/day for 30 days) Experimental Results: diminished levels of the pro-inflammatory cytokines IL-1β and TNF-α. diminished the content of MDA and increased the activity of SOD. Dramatically mitigated D-Gal-induced downregulation of SIRT1 in hippocampal neurons. Dramatically inhibited the expression of p53, acetyl-p53, and p21 in mice (all proteins associated with hippocampal aging). Animal/Disease Models: CAV16-indeced ICR suckling mice model[2] Doses: 2 mg/kg Route of Administration: intramuscular (im) injection; Single dose. After CVA16 treatment (10[5.5] TCID50/g; intramuscular (im) injection; Single dose ) Experimental Results: Delayed death of the mice (6 days post-infection and 7 dpi), and eventually resulted in a survival rate of 50% and 70% for |
| 参考文献 |
[1]. Park SH, et al. Identification of acetylshikonin as the novel CYP2J2 inhibitor with anti-cancer activity in HepG2 cells. Phytomedicine. 2017 Jan 15;24:134-140.
[2]. Shon JC, et al. Acetylshikonin is a novel non-selective cytochrome P450 inhibitor. Biopharm Drug Dispos. 2017 Dec;38(9):553-556. [3]. Wang Y, et al. Acetylshikonin, a Novel AChE Inhibitor, Inhibits Apoptosis via Upregulation of Heme Oxygenase-1 Expression in SH-SY5Y Cells. Evid Based Complement Alternat Med. 2013;2013:937370. |
| 其他信息 |
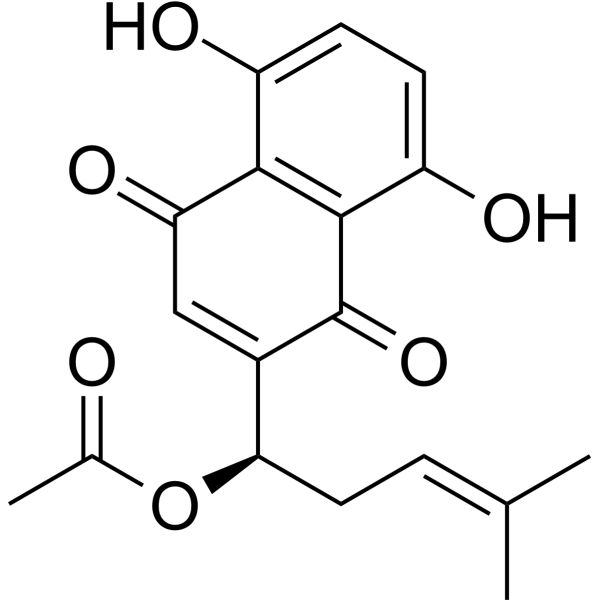
Acetylshikonin is an acetate ester and a hydroxy-1,4-naphthoquinone.
Acetylshikonin has been reported in Arnebia decumbens, Arnebia euchroma, and other organisms with data available. |
| 分子式 |
C18H18O6
|
|---|---|
| 分子量 |
337.3751
|
| 精确质量 |
330.11
|
| CAS号 |
24502-78-1
|
| 相关CAS号 |
DL-Acetylshikonin;54984-93-9
|
| PubChem CID |
32464
|
| 外观&性状 |
Brown to khaki solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
553.2±50.0 °C at 760 mmHg
|
| 熔点 |
86°C
|
| 闪点 |
201.3±23.6 °C
|
| 蒸汽压 |
0.0±1.6 mmHg at 25°C
|
| 折射率 |
1.602
|
| LogP |
4.57
|
| tPSA |
100.9
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
24
|
| 分子复杂度/Complexity |
599
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
WNFXUXZJJKTDOZ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C18H18O6/c1-9(2)4-7-15(24-10(3)19)11-8-14(22)16-12(20)5-6-13(21)17(16)18(11)23/h4-6,8,15,20-21H,7H2,1-3H3
|
| 化学名 |
[1-(5,8-dihydroxy-1,4-dioxonaphthalen-2-yl)-4-methylpent-3-enyl] acetate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~151.36 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (7.57 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9640 mL | 14.8201 mL | 29.6402 mL | |
| 5 mM | 0.5928 mL | 2.9640 mL | 5.9280 mL | |
| 10 mM | 0.2964 mL | 1.4820 mL | 2.9640 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 匹伐加宾
匹伐加宾
 磷酸氢化可的松
磷酸氢化可的松
 BAY-784
BAY-784
 Gly-PEG3-endo-BCN
Gly-PEG3-endo-BCN
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























