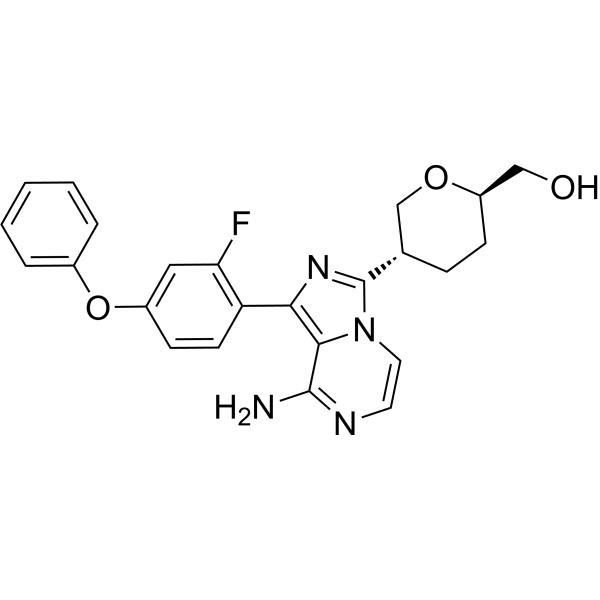
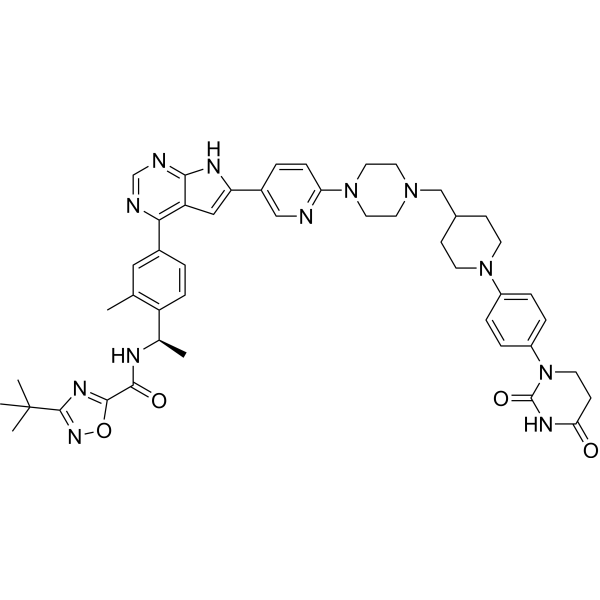
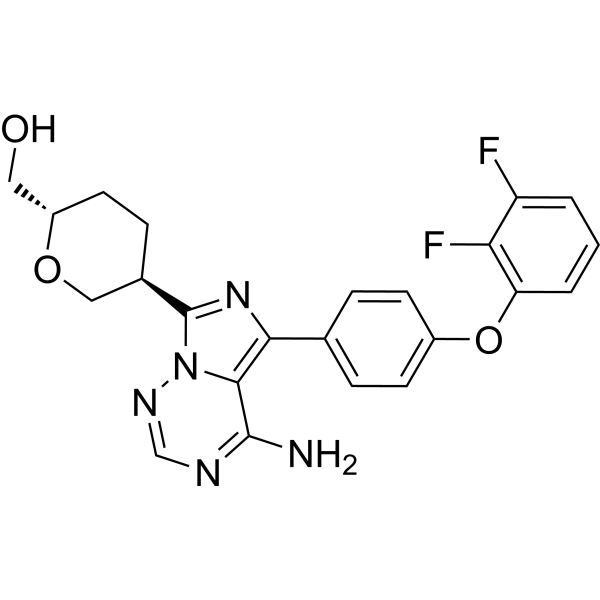
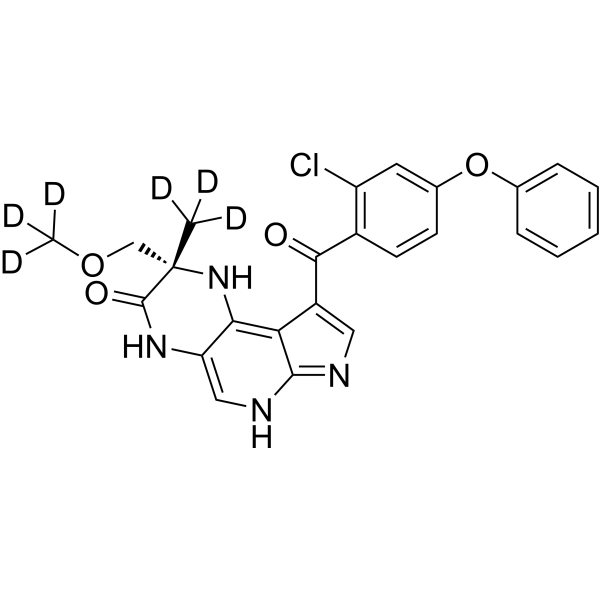
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|
| 靶点 |
BTK (IC50 = 3 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:在原代人 CLL 细胞的体外信号传导测定中,acalabrutinib 抑制 ERK、IKB 和 AKT 下游靶标的酪氨酸磷酸化。 Acalabrutinib 对 BTK 具有更高的选择性,对 9 种具有与 BTK 相同位置的半胱氨酸残基的激酶进行 IC50 测定。重要的是,与依鲁替尼不同,acalabrutinib 不会抑制 EGFR、ITK 或 TEC。 acalabrutinib 对 EGFR 酪氨酸残基 Y1068 和 Y1173 磷酸化没有影响。与 ibrutinib 相比,acalabrutinib 对 ITK、EGFR、ERBB2、ERBB4、JAK3、BLK、FGR、FYN、HCK、LCK、LYN、SRC 和 YES1 激酶活性具有更高的 IC50(>1000 nM)或几乎没有抑制作用。激酶测定:先前的研究表明,当针对一组 395 个非突变激酶进行测试时,ACP-196 对 Btk 具有高选择性,这与 ACP-196 亲电子试剂的内在反应性降低有关。此外,ACP-196 不能抑制 EGFR、Itk 或 Txk,这与依鲁替尼不同。此外,对 EGFR 表达细胞系的磷流分析进一步证实了依鲁替尼对 EGFR 的抑制作用,而没有观察到 ACP-196 的抑制作用。细胞测定:对 EGFR 表达细胞系的磷流测定进一步证实了依鲁替尼对 EGFR 的抑制作用,而没有观察到 ACP-196 的抑制作用。
|
| 体内研究 (In Vivo) |
小鼠口服 ACP-196 会导致 CD19+ 脾细胞中抗 IgM 诱导的 CD86 表达受到剂量依赖性抑制,ED50 为 0.34 mg/kg,而依鲁替尼为 0.91 mg/kg。使用类似的模型来比较单次口服剂量 25 mg/kg 后 Btk 抑制的持续时间。 ACP-196 在给药后 3 小时抑制 CD86 表达 >90%。
Acalabrutinib (ACP-196)的临床前研究[1] Acalabrutinib/阿卡拉布替尼在几种B细胞非霍奇金淋巴瘤(NHL)动物模型中进行了评估。这些研究为阿卡拉布替尼进入人体试验提供了必要的临床前体内数据。在犬B细胞NHL模型研究中,12只B细胞NHL犬口服阿卡拉布替尼,剂量逐渐递增,每24小时2.5 mg/kg(6只狗),每24小时5 mg/kg(5只狗),或每12小时10 mg/kg(1只狗)。结果,3只狗获得部分缓解(PR), 3只狗病情稳定(SD),而其余6只狗病情进展(PD)。因此,本研究表明阿卡拉布替尼在自发性NHL大型动物模型中具有单药生物活性。[1] 阿卡拉布替尼对CLL细胞的体内作用在移植人CLL细胞的NSG小鼠模型中得到了证实。阿卡拉布替尼在所有剂量水平下均能显著抑制NSG小鼠脾脏中人CLL细胞的增殖,检测Ki67的表达(P = 0.002)。阿卡拉布替尼治疗后肿瘤负荷呈剂量依赖性下降。阿卡拉布替尼通过降低plc - γ - 2的磷酸化来抑制BCR信号传导。阿卡拉布替尼可瞬间增加外周血CLL细胞计数。因此,新型BTK抑制剂acalabrutinib对移植到NSG小鼠模型的人CLL细胞具有体内抑制作用。 < br > < br > 在另一项体内研究中使用了两只小鼠模型。在TCL1过继性转移模型中,阿卡拉布替尼通过降低BTK的自磷酸化和降低BCR激活标志物CD86和CD69的表面表达来抑制BCR信号传导。最有趣的是,阿卡拉布替尼治疗显著提高了小鼠的生存率(中位81天vs 59天,P = 0.02)。第二种小鼠模型为NSG异种移植模型。阿卡拉布替尼显著降低了plc - γ - 2和ERK的磷酸化水平(P = 0.02),降低了肿瘤细胞的增殖(P = 0.02),降低了肿瘤负荷(P = 0.04)。Acalabrutinib在两种小鼠CLL模型中均显示为BTK的有效抑制剂。 |
| 酶活实验 |
根据之前的一项研究,当针对一组 395 个非突变激酶进行测试时,ACP-196 的亲电子试剂固有反应性降低与其对 Btk 的高选择性有关。与依鲁替尼相反,ACP-196 无法抑制 EGFR、Itk 或 Txk。通过对表达 EGFR 的细胞系进行磷酸流分析,进一步证实了依鲁替尼在不抑制 ACP-196 的情况下抑制 EGFR 的作用。
与ibrutinib和CC-292相比,ACP-196在竞争结合实验中对395种非突变激酶(1 μM)进行了分析,显示出更高的Btk选择性。对与Btk相同位置的9个激酶的IC50测定表明,ACP-196的选择性最强。选择性的提高与ACP-196亲电试剂的本征反应性降低有关。重要的是,与依鲁替尼不同,ACP-196不抑制EGFR、Itk或Txk。在表达EGFR的细胞株上进行的磷酸流实验证实了依鲁替尼对EGFR的抑制作用(EC50: 47 ~ 66 nM),而在10 μM下对ACP-196没有抑制作用。这些数据可以解释依鲁替尼相关的腹泻和皮疹发生率。伊鲁替尼对Itk和Txk的效力可能解释了为什么它会干扰治疗性CD20抗体的细胞介导的抗肿瘤活性和肿瘤微环境中免疫介导的杀伤。 在人全血中,ACP-196和伊鲁替尼在低nM范围内对b细胞受体诱导的反应表现出强大的、同等的抑制活性,而CC-292的抑制作用弱10-20倍。[3] |
| 细胞实验 |
磷酸流测试中使用的表达 EGFR 的细胞系进一步验证了依鲁替尼对 EGFR 的抑制作用,未观察到任何 ACP-196 抑制作用。
最近认识到b细胞受体(BCR)信号是b细胞恶性肿瘤(包括非霍奇金淋巴瘤(NHL))进展的关键因素,导致许多靶向治疗方法的发展,抑制这一信号通路。Ibrutinib是一种布鲁顿酪氨酸激酶(Btk)的小分子抑制剂,Btk是BCR通路中的关键信号分子,已在广泛的b细胞癌中显示出显着的临床活性。ACP-196是第二代Btk抑制剂,与伊鲁替尼相比,具有更高的靶标选择性和更高的体内效价,因此可能比其前身有所改进。在接下来的研究中,我们试图评估ACP-196在犬b细胞NHL模型中的作用,最终目的是为ACP-196进入人体临床试验提供必要的临床前数据。利用两种免疫表型证实的犬b细胞淋巴瘤细胞系CLBL-1和17-71,我们证明了ACP-196在低至10nM的浓度下治疗1小时后,对Btk和下游效应物ERK 1/2和PLCγ2的激活有有效的体外抑制作用。[2] |
| 动物实验 |
canine model of B cell NHL
2.5, 5, 10 mg/kg. orally administered In vivo studies were performed in companion dogs as part of an ongoing clinical trial. Twelve dogs with immunophenotypically confirmed, spontaneously occurring B-cell NHL were orally administered ACP-196 at dosages of 2.5mg/kg every 24 hours (6 dogs), 5mg/kg every 24 hours (5 dogs), or 10mg/kg every12 hours (1 dog). Btk occupancy in peripheral blood and lymphoma cells was assessed using a biotin-tagged probe derived from ACP-196. Using this assay we found that at 2.5mg/kg full Btk occupancy was achieved in peripheral B cells 3h after dosing for all dogs, except for a single dog with high peripheral B-cell count. At 24 hours after dosing, 83-99% Btk target occupancy was observed for all dogs. Partial response, as assessed by a modified RECIST scheme, was achieved in 2 dogs in the 2.5mg/kg group and the dog in the 10mg/kg group. Upon relapse, one of the responders in the 2.5mg/kg group was dose escalated to 10mg/kg q12 on day 42 and partial response from relapse was reestablished. Of the remaining 9 dogs, 3 achieved stable disease for > 28 days and 6 discontinued the study after developing progressive disease within 28 days of starting treatment. In total, to date, 3 dogs achieved a partial response, 3 dogs stable disease, and 6 dogs progressive disease. ACP-196 was well tolerated with only mild anorexia noted in some dogs. These data demonstrate that ACP-196 has single agent biologic activity in a spontaneous large animal model of human NHL. Studies in dogs with NHL are ongoing to define regimens prior to initiation of human phase I clinical trials. Additional cohorts are planned combining ACP-196 with a phosphatidylinositide 3-kinase (PI3K) delta-specific inhibitor.[2] In vivo, oral administration of ACP-196 in mice resulted in dose-dependent inhibition of anti-IgM-induced CD86 expression in CD19+ splenocytes with an ED50 of 0.34 mg/kg compared to 0.91 mg/kg for ibrutinib. A similar model was used to compare the duration of Btk inhibition after a single oral dose of 25 mg/kg. ACP-196 and ibrutinib inhibited CD86 expression >90% at 3h and ∼50% at 24h postdose. In contrast, CC-292 inhibited ∼50% at 3h and ∼20% at 24h postdose. An ELISA based Btk target occupancy assay was developed to measure target coverage in preclinical and clinical studies. In healthy volunteers, ACP-196 at an oral dose of 100 mg QD showed >90% target coverage over a 24h period. Btk occupancy and regulation of the PD markers (CD69 and CD86) correlated with PK parameters for exposure. In CLL patients, after 7 days of dosing with ACP-196 at 200 mg QD, 94% Btk target occupancy was observed compared with ∼80% reported for ibrutinib at 420 mg QD [3]. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
The geometric mean absolute bioavailability of acalabrutinib is 25% with a median time to peak plasma concentrations (Tmax) of 0.75 hours. After administration of a single 100 mg radiolabelled acalabrutinib dose in healthy subjects, 84% of the dose was recovered in the feces and 12% of the dose was recovered in the urine. An irradiated dose of acalabrutinib was 34.7% recovered as the metabolite ACP-5862; 8.6% was recovered as unchanged acalabrutinub; 10.8 was recovered as a mixture of the M7, M8, M9, M10, and M11 metabolites; 5.9% was the M25 metabolite; 2.5% was recovered as the M3 metabolite. The mean steady-state volume of distribution is approximately 34 L. Acalabrutinib's mean apparent oral clearance (CL/F) is observed to be 159 L/hr with similar PK between patients and healthy subjects, based on population PK analysis. Metabolism / Metabolites Acalabrutinib is mainly metabolized by CYP3A enzymes. ACP-5862 is identified to be the major active metabolite in plasma with a geometric mean exposure (AUC) that is about 2-3 times greater than the exposure of acalabrutinib. ACP-5862 is about 50% less potent than acalabrutinib in regards to the inhibition of BTK. Biological Half-Life After administering a single oral dose of 100 mg acalabrutinib, the median terminal elimination half-life of the drug was found to be 0.9 (with a range of 0.6 to 2.8) hours. The half-life of the active metabolite, ACP-5862, is about 6.9 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In open label clinical trials of acalabrutinib in patients with CLL and mantle cell lymphoma, serum aminotransferase elevations occurred in 19% to 23% of patients during therapy and rose to above 5 times ULN in 2% to 3%. These elevations were transient and resolved spontaneously but occasionally led to early drug discontinuation. Among the 610 patients treated with acalabrutinib in pre-registration trials, there were no instances of clinically apparent liver injury attributed to its use, but there was a single instance of acute liver failure and death due to reactivation of hepatitis B. Similar cases of reactivation have been reported with ibrutinib, another small molecule inhibitor of Bruton's tyrosine kinase. Experience with acalabrutinib has been limited and the frequency of clinically apparent liver injury and reactivation of hepatitis B are not known. The majority of cases have occurred in patients taking multiple immunosuppressive agents and not just acalabrutinib alone. Likelihood score: D (possible rare cause of reactivation of hepatitis B). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of acalabrutinib during breastfeeding. Because acalabrutinib is over 97% bound to plasma proteins, and the half-life of the drug and metabolite are less than 7 hours, the amount in milk is likely to be low. However, the protein binding of the active metabolite is not known and the manufacturer recommends that breastfeeding be discontinued during acalabrutinib therapy and for at least 2 weeks after the final dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Reversible binding of acalabrutinib to human plasma protein is approximately 97.5%. The in vitro mean blood-to-plasma ratio is about 0.7. _In vitro_ experiments at physiologic concentrations show that acalabrutinib can be 93.7% bound to human serum albumin and 41.1% bound to alpha-1-acid glycoprotein. |
| 参考文献 | |
| 其他信息 |
Pharmacodynamics
Acalabrutinib is a Bruton Tyrosine Kinase inhibitor that prevents the proliferation, trafficking, chemotaxis, and adhesion of B cells. It is taken every 12 hours and can cause other effects such as atrial fibrillation, other malignancies, cytopenia, hemorrhage, and infection. |
| 分子式 |
C26H23N7O2
|
|
|---|---|---|
| 分子量 |
465.51
|
|
| 精确质量 |
465.191
|
|
| 元素分析 |
C, 67.08; H, 4.98; N, 21.06; O, 6.87
|
|
| CAS号 |
1420477-60-6
|
|
| 相关CAS号 |
Acalabrutinib-d4;2699608-18-7;Acalabrutinib-d3; 1420477-60-6; 2058091-99-7 (citrate); 2242394-65-4; 2058091-96-4 (phosphate); 2058091-93-1 (3 hydrate); 2058092-05-8 (sulfate); 2058091-94-2 (fumarate); 2058091-97-5 (tartrate)
|
|
| PubChem CID |
71226662
|
|
| 外观&性状 |
Yellow solid powder
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 折射率 |
1.715
|
|
| LogP |
0.77
|
|
| tPSA |
118.51
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
35
|
|
| 分子复杂度/Complexity |
845
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
O=C(C#CC([H])([H])[H])N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C1=NC(C2C([H])=C([H])C(C(N([H])C3=C([H])C([H])=C([H])C([H])=N3)=O)=C([H])C=2[H])=C2C(N([H])[H])=NC([H])=C([H])N12
|
|
| InChi Key |
WDENQIQQYWYTPO-IBGZPJMESA-N
|
|
| InChi Code |
InChI=1S/C26H23N7O2/c1-2-6-21(34)32-15-5-7-19(32)25-31-22(23-24(27)29-14-16-33(23)25)17-9-11-18(12-10-17)26(35)30-20-8-3-4-13-28-20/h3-4,8-14,16,19H,5,7,15H2,1H3,(H2,27,29)(H,28,30,35)/t19-/m0/s1
|
|
| 化学名 |
4-[8-amino-3-[(2S)-1-but-2-ynoylpyrrolidin-2-yl]imidazo[1,5-a]pyrazin-1-yl]-N-pyridin-2-ylbenzamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.47 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.47 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (4.47 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2% DMSO+30% PEG 300+2% Tween 80+ddH2O: 6mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1482 mL | 10.7409 mL | 21.4818 mL | |
| 5 mM | 0.4296 mL | 2.1482 mL | 4.2964 mL | |
| 10 mM | 0.2148 mL | 1.0741 mL | 2.1482 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04008706 | Active Recruiting |
Drug: Acalabrutinib | Chronic Lymphocytic Leukemia | AstraZeneca | September 17, 2019 | Phase 3 |
| NCT05256641 | Recruiting | Drug: Acalabrutinib | High-grade B-cell Lymphoma Transformed Lymphoma |
Jonsson Comprehensive Cancer Center |
January 23, 2023 | Phase 1 Phase 2 |
| NCT05951959 | Not yet recruiting | Drug: Acalabrutinib Drug: Venetoclax |
Mantle Cell Lymphoma (MCL) |
AstraZeneca | November 9, 2023 | Phase 2 |
| NCT05004064 | Not yet recruiting | Drug: Acalabrutinib Drug: Rituximab |
Mantle Cell Lymphoma | University College, London | January 1, 2023 | Phase 2 |
| NCT04402138 | Active Recruiting |
Drug: Acalabrutinib | Mantle Cell Lymphoma | SCRI Development Innovations, LLC |
August 7, 2020 | Phase 2 |
Acalabrutinib demonstrates equalin vitroon-target effects as ibrutinib.Clin Cancer Res.2017 Jun 1;23(11):2831-2841. |
|---|
Acalabrutinib demonstrates on target effects and reduced proliferation and tumor burden in the CLL xenograft mouse model.Clin Cancer Res.2017 Jun 1;23(11):2831-2841. |
Acalabrutinib demonstrates significant and sustained inhibition of BCR signaling in the TCL1 adoptive transfer model.Clin Cancer Res.2017 Jun 1;23(11):2831-2841. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
")
")
")