| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 100mg |
|

| 靶点 |
wild-type ROS1; ROS1 G2032R; ROS1 fusions and resistance mutants
|
|---|---|
| 体外研究 (In Vitro) |
Zidesamtinib (72 h) 的平均 IC50 为 0.4 nM,抑制表达野生型 ROS1 融合的七种细胞系的生长[1]。
Zidesamtinib (72 h) 的平均 IC50 为 1.6 nM,抑制六种带有 G2032R 突变的 ROS1 融合细胞系[1]。 Zidesamtinib (72 h) 有效抑制非 G2032R ROS1 突变体,IC50 ≤ 1.5 nM[1]。 Zidesamtinib (10-1000 nM; 4 周)抑制 NIH3T3 细胞中表达 ROS1 与 G2032R 融合和野生型 ROS1 融合的集落形成[1]。 |
| 体内研究 (In Vivo) |
Zidesamtinib(0.04-15 mg/kg;每天口服两次,持续 28 天)在所有剂量≥0.2 mg/kg 的野生型 ROS1 异种移植模型中都能促进肿瘤消退[1]。
|
| 酶活实验 |
生化激酶活性测定[1]
使用PhosphoSens测定法测量纯化激酶的活性。将测试化合物溶解在DMSO中至所需浓度的100倍,并在250nL下以3倍稀释系列分配到384孔板中。将含有2 mmol/L ATP的12.5μL溶液与26μmol/L荧光肽底物AQT0101或AQT0104的缓冲液(50 mmol/L HEPES pH 7.5,0.01%Brij-35,0.5 mmol/L EGTA,10 mmol/L MgCl2)加入板中。通过加入12.5μL溶液引发反应,该溶液在缓冲液中含有0.5 nmol/L ROS1、2 nmol/L ROS 1 G2032R、1 nmol/L TRKA、3 nmol/L TR KB或0.5 nmol/L TRKC激酶结构域(50 mmol/L HEPES pH 7.5,0.01%Brij-35,2%甘油,0.4 mg/mL BSA,0.5 mmol/L EGTA,10 mmol/L MgCl2)。最终浓度为1 mmol/L ATP、13μmol/L肽底物(ROS1和ROS1 G2032R的AQT0101或TRKA、TRKB和TRKC的AQT0104)、0.25至1.5 nmol/L激酶(0.25 nmol/L ROS1;1 nmol/L ROS 1 G2032 R;0.5 nmol/L TRKA;1.5 nmol/L TRKB;或0.5 nmol/L TRKC)、50 mmol/L HEPES pH 7.5、0.01%Brij-35、1%甘油、0.2 mg/mL BSA、0.5 mmol/L EGTA和10 mmol/L MgCl2。将平板密封,在30°C下,用平板阅读器在λ发射=485nm下每2分钟记录荧光信号120分钟。在反应的初始线性阶段,荧光强度随时间的变化是初始速度(v)。IC50由初始速度与对数(抑制剂浓度)回归到四参数逻辑斯谛方程的图计算得出。[1] 激酶面板筛查[1] 使用放射性标记的[γ-33P]-ATP测量激酶活性的抑制。将含有[γ-33P]-ATP的溶液与NVL-520、335种激酶中的每一种以及相应的激酶底物混合。NVL-520在两种浓度下进行了测定:100 nmol/L和1μmol/L。该测定中ATP的浓度接近每种激酶的米氏常数(KM)。1小时后停止反应,使用闪烁计数器定量33P的掺入。通过33P的残余活性来衡量抑制作用:较低的残余活性表明特定激酶的抑制剂效力较高。基于335激酶筛选,选择24种抑制率>50%的激酶,使用相同的方法在10个浓度的NVL-520下以半对数步骤(3μmol/L、900 nmol/L、300 nmol/L和90 nmol/L,…、0.09 nmol/L)进行IC50测定。IC50由残余活性与对数(抑制剂浓度)回归到四参数逻辑斯谛方程的图确定。 |
| 细胞实验 |
细胞活力测定[1]
协议因测试地点而异。对于测试部位A,将A549或稳定的Ba/F3细胞接种到384孔板中,并在含有10%FBS的完整培养基中以3倍稀释系列加入测试化合物。用抑制剂孵育72小时后,使用CellTiter-Glo试剂测量细胞存活率。未处理的孔作为阴性对照(不抑制增殖),而用高浓度非特异性激酶抑制剂星孢菌素处理的孔则作为阳性对照(完全抑制增殖)。使用四参数逻辑回归从抑制百分比和log(抑制剂浓度)计算IC50。[1] 对于测试部位B,所有抑制剂均以1 mmol/L的DMSO储备制备。使用多点组合试剂分配器,将每孔25μL的完整培养基预先接种到平板上。使用D300数字分配器将抑制剂以指示浓度的2倍分配到384孔板上,每孔25μL的完整培养基中。使用多点组合试剂分配器以每孔1000个细胞的体积25μL接种表达野生型或突变型ROS1融合物的Ba/F3细胞系。将板温育72小时。使用基于WST-8[2-(2-甲氧基-4-硝基苯基)-3-(4-硝基苯基)-5-(2,4-二磺苯基)-2H-四唑鎓单钠盐]的测定法测量存活率,并在Biotek Synergy 2平板读数器上读取。每种情况都进行了三次检测。使用Microsoft Excel对数据进行归一化,并使用GraphPad Prism中的非线性回归分析计算IC50值。[1] 对于测试部位C,将MGH193-1和MGH9018-1细胞以4000个细胞/孔的速度一式三份铺在96孔板上。经过5天的药物处理后,用CellTiter Glo孵育细胞,并用SpectraMax M5多模微孔板读数器测量发光。GraphPad Prism(GraphPad软件)用于图形化显示数据,并通过使用四参数分析方法的非线性回归模型确定IC50值。[1] 在一些人类癌症细胞系中,我们观察到NVL-520表现出双相剂量-反应行为。我们将第一次剂量反应归因于ROS1抑制引起的靶向生长抑制,将第二次剂量反应归咎于ROS1抑制剂以外途径引起的脱靶细胞毒性。在这种情况下,只报告了第一个IC50值,并指示对靶ROS1的抑制。[1] 细胞磷酸化测定[1] 对于Ba/F3 TRKB细胞磷酸化测定,将细胞接种到384孔板中,并在全培养基+10%FBS中以3倍稀释系列加入测试化合物。用100 ng/mL BDNF刺激细胞20分钟。使用磷酸化TRKA(Tyr674/675)/磷酸化TRKB(Tyr706/Tyr707)AlphaLISA试剂测量TRK磷酸化。未处理的孔作为阴性对照(无抑制),而用高浓度非特异性激酶抑制剂星孢菌素处理的孔则作为阳性对照(完全抑制)。使用四参数逻辑回归从抑制百分比和抑制剂浓度计算IC50。[1] 表达EZR-ROS1野生型或突变型融合物的NIH3T3细胞在收获前用指定浓度的抑制剂处理3小时。用磷酸盐缓冲盐水(PBS)洗涤细胞,用补充有0.25%脱氧胆酸盐、0.05%SDS和蛋白酶和磷酸酶抑制剂的细胞裂解缓冲液收获细胞。使用皮尔斯BCA蛋白测定法测定蛋白质浓度。使用添加了β-巯基乙醇的Laemelli样品缓冲液在75°C下提取裂解物10分钟,裂解物在4%至20%的预制梯度Bis-tris凝胶上运行。[1] MGH193-1和MGH9018-1处理6小时。用以下抗体通过蛋白质印迹分析总蛋白裂解物:磷酸-ROS1 Y2274(3078)、ROS1(3287)、磷酸AKT S473(4060)、AKT(4691)、磷酸-ERK1/2 T202/Y204(9101)、ERK1/2(9102)、磷酸-S6 S240/244(5364)、S6(2217)和β-Actin(4970)。[1] 菌落形成试验[1] 在含有DMSO或抑制剂(10、100或1000 nmol/L的克唑替尼、恩替尼、洛拉替尼或NVL-520)的完全培养基中,用0.8%琼脂糖预接种平板。每种抑制剂都与自己的DMSO条件配对,作为精确的对照。将表达CD74-ROS1或EZR-ROS1野生型或突变型融合的NIH3T3细胞以每0.5 mL琼脂糖2000个细胞的密度,用与底层相同浓度的DMSO或抑制剂,在完全培养基中的0.4%琼脂糖中铺板。培养板孵育4周,每孔每周3次喂食75μL含或不含抑制剂的完全培养基,以匹配每种培养条件,防止琼脂糖干燥。3周和4周后使用GelCount读取板。菌落计数按条件平均,并归一化为成对DMSO条件下的菌落计数。使用Microsoft Excel和GraphPad Prism进行数据分析和可视化。 |
| 动物实验 |
Female athymic Nude-Foxn1nu mice were implanted subcutaneously with tumor fragments from model CTG-0848[1]
0.04, 0.2, 1, 5, 15 mg/kg Oral gavage twice daily for 21 days Subcutaneous Xenograft Studies.[1] CTG-0848 PDX. [1] The experimental procedures were performed according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Champions Oncology, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Female athymic Nude-Foxn1nu mice were implanted subcutaneously into the left flank with tumor fragments from the CTG-0848 model. In the efficacy study, after tumors grew to 150 to 300 mm3, mice (n = 3–5/group) were randomized and administered vehicle or NVL-520 by oral gavage b.i.d. (12-hour intervals). In a separate pharmacokinetic (PK) and pharmacodynamic (PD) study, after tumors grew to 350 to 500 mm3, mice received a single dose or b.i.d. × 5 days of vehicle or NVL-520, and tumor and blood were collected at 1 hour and 12 hours (treatment only) after dose. The model was verified by vendor-provided NGS to contain a heterozygous CD74–ROS1 fusion.[1] View More
Lu01-0414 PDX.[1] MGH9018-1 PDX. [1] The study was conducted in accordance with the guidelines as published in the Guide for the Care and Use of Laboratory Animals and was approved by the IACUC of Massachusetts General Hospital. Xenografts were implanted subcutaneously into the flanks of female athymic nude (Nu/Nu) mice ages 6 to 8 weeks. Mice were maintained in laminar flow units in sterile filter-top cages with Alpha-Dri bedding. Mice were randomized into groups once the tumors had attained a volume of 150 mm3. The treatment groups were treated with drug solution dissolved in acid water once a day (crizotinib) or drug solution dissolved in 20% hydroxypropyl-b-cyclodextrin (HP-β-CD) twice a day at 9/15-hour intervals (NVL-520) by oral gavage. Tumor volumes were measured twice weekly and calculated using the formula: mm3 = 0.52 × L × W2. For protein assays, tumor-bearing mice were administered drugs for 3 days according to the above dosing schedule, and tumors were harvested 3 hours after the last treatment for Western blotting.[1] CTG-2532 PDX. [1] The experimental procedures were performed according to the guidelines approved by the IACUC of Champions Oncology, accredited by AAALAC. Female athymic Nude-Foxn1nu mice were implanted subcutaneously into the left flank with tumor fragments from model CTG-2532. In the efficacy study, after tumors grew to 150 to 300 mm3, mice (n = 5/group) were administered vehicle, NVL-520, or repotrectinib by oral gavage b.i.d. (12-hour intervals) for up to 21 days (b.i.d. × 21 days) if tolerated. Repotrectinib (DC Chemicals) was dosed as a suspension solution containing 0.5% carboxymethylcellulose and 1% Tween-80. The dosing suspension was stored 2–8°C in the dark for up to 7 days with continuous stirring. In a separate PK and PD study, CTG-2532 tumors were subcutaneously implanted in mice and allowed to grow to 350 to 550 mm3 before a single dose of the vehicle or NVL-520 was administered. Tumor and blood were collected at 1 hour and 12 hours (treatment only) after dose for PK and PD analysis. The model was verified by vendor-provided NGS to harbor a CD74–ROS1 G2032R. Ba/F3 CD74–ROS1 G2032R CDX. All the procedures related to animal handling, care, and treatment in this study were performed according to guidelines approved by the IACUC of Pharmaron following the guidance of the AAALAC. Six- to 8-week-old female Balb/c nude mice were inoculated subcutaneously on the right flank with 1 × 106 Ba/F3 CD74–ROS1 G2032R cells. After the tumors reached a mean tumor volume of approximately 128 mm3, treatment was initiated, and tumors and body weight were measured at regular intervals.[1] Plasma drug concentrations were determined by LC/MS. Free drug concentrations were calculated by multiplying the total concentration values determined from PK experiments with the corresponding fraction unbound in mouse plasma (= 7.7% for NVL-520 and = 7.6% for crizotinib).[1] Kp,uu and Intracranial Studies[1] All the procedures related to animal handling, care, and treatment in these studies were performed according to guidelines approved by the IACUC of Pharmaron following the guidance of AAALAC.[1] NVL-520 was formulated as 1 mg/mL suspension in 20% HP-β-CD in deionized water. Lorlatinib was formulated as a 1 mg/mL solution in two equivalents of HCl + 20% HP-β-CD in deionized water. Compounds were administered orally to male Wistar Han rats (n = 3 each). After 1 hour, brain samples and plasma were collected, and brain samples were homogenized in PBS. Brain and plasma samples were precipitated by acetonitrile and centrifugation (4,700 rpm, 15 minutes). Drug concentrations in the supernatants were quantified by LC/MS-MS. Unbound fractions were determined using rapid equilibrium dialysis. Kp,uu was calculated as the ratio between unbound drug concentration in the brain and unbound drug concentration in the plasma.[1] Ba/F3 CD74–ROS1 G2032R cells were transduced with viral particles containing the firefly luciferase gene and a neomycin resistance marker. Infected cells were selected on neomycin, and monoclonal cultures were established through limiting dilution. Successful transformants were confirmed by Sanger sequencing and bioluminescence. For the in vivo study, 1 × 105 Ba/F3 CD74–ROS1 G2032R luciferase cells were stereotactically implanted into the right forebrains of 6- to 8-week-old female Balb/c nude mice. After 5 days, mice were randomized based on mean bioluminescence signal into three groups of n = 7–10 mice each and received vehicle or NVL-520, 2 mg/kg, orally, b.i.d. Bioluminescence and body weight were measured at regular intervals until the end of the study (61 days after treatment start) or until animals met the criteria for euthanasia. |
| 参考文献 | |
| 其他信息 |
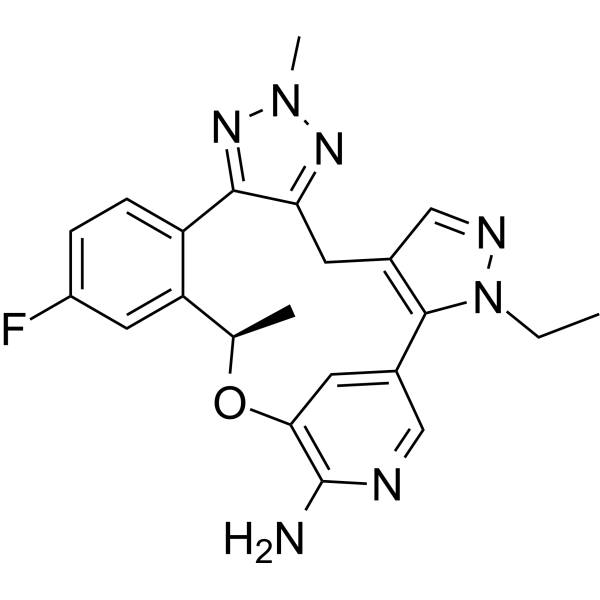
Zidesamtinib is an orally available selective inhibitor of the receptor tyrosine kinase c-ros oncogene 1 (ROS1), with potential antineoplastic activity. Upon oral administration, zidesamtinib targets, binds to and inhibits wild-type, point mutants and fusion proteins of ROS1. This inhibits proliferation of ROS-1-driven tumor cells, including in tumor cells harboring certain ROS1 resistance mutations, such as the solvent front mutation G2032R and the S1986Y/F, L2026M, and D2033N resistance mutations. Inhibition of ROS1 leads to the disruption of downstream signaling pathways and the inhibition of cell growth of tumors in which ROS1 overexpressed, rearranged or mutated. ROS1, overexpressed in certain cancer cells, plays a key role in cell growth and survival of cancer cells. NVL-520 is able to penetrate the blood-brain barrier (BBB).
The combined preclinical features of NVL-520 that include potent targeting of ROS1 and diverse ROS1 resistance mutations, high selectivity for ROS1 G2032R over TRK, and brain penetration mark the development of a distinct ROS1 TKI with the potential to surpass the limitations of earlier-generation TKIs for ROS1 fusion-positive patients.[1] |
| 分子式 |
C22H22FN7O
|
|---|---|
| 精确质量 |
419.46
|
| 元素分析 |
C, 63.00; H, 5.29; F, 4.53; N, 23.37; O, 3.81
|
| CAS号 |
2739829-00-4
|
| 相关CAS号 |
2739829-00-4
|
| PubChem CID |
166560233
|
| 外观&性状 |
Typically exists as white to off-white solids at room temperature
|
| LogP |
2.8
|
| tPSA |
96.7Ų
|
| SMILES |
CCN1C2=C(CC3=NN(N=C3C4=C(C=C(C=C4)F)[C@H](OC5=C(N=CC2=C5)N)C)C)C=N1
|
| InChi Key |
DTWUUAFTYSMNQX-GFCCVEGCSA-N
|
| InChi Code |
InChI=1S/C22H22FN7O/c1-4-30-21-13(11-26-30)7-18-20(28-29(3)27-18)16-6-5-15(23)9-17(16)12(2)31-19-8-14(21)10-25-22(19)24/h5-6,8-12H,4,7H2,1-3H3,(H2,24,25)/t12-/m1/s1
|
| 化学名 |
(19R)-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,9,10,11,23-hexazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine
|
| 别名 |
NVL520; NVL 520; NVL-520; Zidesamtinib; NVL-520; 2739829-00-4; NVL520; zidesamtinib [INN]; MX5KQV5XHC; Zidesamtinib [WHO-DD]; ZIDESAMTINIB [USAN];
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~119.20 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 12.5 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 12.5 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
 ROS1-IN-2
ROS1-IN-2
 PRDX1-IN-1
PRDX1-IN-1
 Keap1-Nrf2-IN-11
Keap1-Nrf2-IN-11
 Taletrectinib adipate (DS-6051b; AB-106; IBI-344)
Taletrectinib adipate (DS-6051b; AB-106; IBI-344)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































