
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| Other Sizes |
|

| 体外研究 (In Vitro) |
药物化合物包括碳、氢和其他元素的稳定重同位素,在药物开发过程中主要作为定量示踪剂。由于氘化可能会影响药物的药代动力学和代谢特性,因此值得关注[1]。
|
|---|---|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Itraconazole is rapidly absorbed after oral administration. As oral capsules, peak plasma concentrations of itraconazole are reached within two to five hours. The observed absolute oral bioavailability of itraconazole is about 55%. Itraconazole exposure is lower with the capsule formulation than with the oral solution when the same dose of the drug is given. Maximal drug absorption is achieved under adequate gastric acidity. As a consequence of non-linear pharmacokinetics, itraconazole accumulates in plasma during multiple dosing. Steady-state concentrations are generally reached within about 15 days, with Cmax values of 0.5 μg/mL, 1.1 μg/mL and 2.0 μg/mL after oral administration of 100 mg once daily, 200 mg once daily and 200 mg b.i.d., respectively. Itraconazole is excreted mainly as inactive metabolites in urine (35%) and in feces (54%) within one week of an oral solution dose. Renal excretion of itraconazole and the active metabolite hydroxyitraconazole account for less than 1% of an intravenous dose. Based on an oral radiolabeled dose, fecal excretion of unchanged drug ranges from 3% to 18% of the dose. As the re-distribution of itraconazole from keratinous tissues appears to be negligible, the elimination of itraconazole from these tissues is related to epidermal regeneration. Contrary to plasma, the concentration in skin persists for two to four weeks after discontinuation of a 4-week treatment and in nail keratin – where itraconazole can be detected as early as one week after the start of treatment – for at least six months after the end of a 3-month treatment period. The volume of distribution is more than 700 L in adults. Itraconazole is lipophilic and extensively distributed into tissues. Concentrations in the lung, kidney, liver, bone, stomach, spleen and muscle were found to be two to three times higher than corresponding concentrations in plasma, and the uptake into keratinous tissues, skin in particular, up to four times higher. Concentrations in the cerebrospinal fluid are much lower than in plasma. The mean total plasma clearance following intravenous administration is 278 mL/min. Itraconazole clearance decreases at higher doses due to saturable hepatic metabolism. The pharmacokinetics of itraconazole after intravenous administration and its absolute oral bioavailability from an oral solution were studied in a randomized crossover study in 6 healthy male volunteers. The observed absolute oral bioavailability of itraconazole was 55%. The oral bioavailability of itraconazole is maximal when itraconazole capsules are taken with a full meal. The pharmacokinetics of itraconazole were studied in 6 healthy male volunteers who received, in a crossover design, single 100 mg doses of itraconazole as a polyethylene glycol capsule, with or without a full meal. The same 6 volunteers also received 50 mg or 200 mg with a full meal in a crossover design. In this study, only itraconazole plasma concentrations were measured. The respective pharmacokinetic parameters for itraconazole are presented in the table /provided/. Table: Oral Bioavailability of Itraconazole (Itraconazole capsules): [Table#7579] Metabolism / Metabolites Itraconazole is extensively metabolized in the liver. In vitro studies have shown that CYP3A4 is the major enzyme involved in the metabolism of itraconazole. While itraconazole can be metabolized to more than 30 metabolites, the main metabolite is hydroxyitraconazole. Hydroxyitraconazole has in vitro antifungal activity comparable to itraconazole; trough plasma concentrations of this metabolite are about twice those of the parent compound. Other metabolites include keto-itraconazole and N-dealkyl-itraconazole. Itraconazole is metabolized predominantly by the cytochrome P450 3A4 isoenzyme system (CYP3A4), resulting in the formation of several metabolites, including hydroxyitraconazole, the major metabolite. Results of a pharmacokinetics study suggest that itraconazole may undergo saturable metabolism with multiple dosing. Itraconazole (ITZ) is metabolized in vitro to three inhibitory metabolites: hydroxy-itraconazole (OH-ITZ), keto-itraconazole (keto-ITZ), and N-desalkyl-itraconazole (ND-ITZ). The goal of this study was to determine the contribution of these metabolites to drug-drug interactions caused by ITZ. Six healthy volunteers received 100 mg ITZ orally for 7 days, and pharmacokinetic analysis was conducted at days 1 and 7 of the study. The extent of CYP3A4 inhibition by ITZ and its metabolites was predicted using this data. ITZ, OH-ITZ, keto-ITZ, and ND-ITZ were detected in plasma samples of all volunteers. A 3.9-fold decrease in the hepatic intrinsic clearance of a CYP3A4 substrate was predicted using the average unbound steady-state concentrations (C(ss,ave,u)) and liver microsomal inhibition constants for ITZ, OH-ITZ, keto-ITZ, and ND-ITZ. Accounting for circulating metabolites of ITZ significantly improved the in vitro to in vivo extrapolation of CYP3A4 inhibition compared to a consideration of ITZ exposure alone. Biological Half-Life The terminal half-life of itraconazole generally ranges from 16 to 28 hours after a single dose and increases to 34 to 42 hours with repeated dosing. The metabolite of itraconazole is excreted from the plasma more rapidly than the parent compound. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Transient, mild-to-moderate elevations in serum aminotransferase levels occur in 1% to 5% of patients on itraconazole. These elevations are largely asymptomatic and self-limited, resolving even with continuation of therapy. Clinically apparent hepatotoxicity is rare but has been well described and can be severe and even fatal. The liver injury from itraconazole typically presents 1 to 6 months after starting therapy with symptoms of fatigue and jaundice. The pattern of serum enzyme elevations is typically cholestatic (Case 1), but cases of severe hepatitis with acute liver failure typically have a hepatocellular enzyme pattern (Case 2). Immunoallergic features (rash, fever, eosinophilia) are uncommon as is autoantibody formation. Recovery upon stopping therapy can be delayed for several weeks and generally takes 4 to 10 weeks, although in some cases recovery may be prolonged. Likelihood score: B (likely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of itraconazole during breastfeeding. However, limited data indicate that maternal itraconazole produces levels in milk that are less than the 5 mg/kg daily doses that have been recommended to treat infants. Until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. If itraconazole is used during breastfeeding, monitoring of the infant’s liver enzymes should be considered, especially with long courses of therapy. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Most of the itraconazole in plasma is bound to protein (99.8%), with albumin being the main binding component (99.6% for the hydroxy-metabolite). It also has a marked affinity for lipids. Only 0.2% of the itraconazole in plasma is present as the free drug. Interactions The class IA antiarrhythmic quinidine and class III antiarrhythmic dofetilide are known to prolong the QT interval. Co-administration of quinidine or dofetilide with itraconazole may increase plasma concentrations of quinidine or dofetilide which could result in serious cardiovascular events. Therefore, concomitant administration of itraconazole and quinidine or dofetilide is contraindicated. The class IA antiarrhythmic disopyramide has the potential to increase the QT interval at high plasma concentrations. Caution is advised when itraconazole and disopyramide are administered concomitantly. Concomitant administration of digoxin and itraconazole has led to increased plasma concentrations of digoxin. Reduced plasma concentrations of itraconazole were reported when itraconazole was administered concomitantly with phenytoin. Carbamazepine, phenobarbital and phenytoin are all inducers of CYP3A4. Although interactions with carbamazepine and phenobarbital have not been studied, concomitant administration of itraconazole and these drugs would be expected to result in decreased plasma concentrations of itraconazole. Drug interaction studies have demonstrated that plasma concentrations of azole antifungal agents and their metabolites, including itraconazole and hydroxyitraconazole, were significantly decreased when these agents were given concomitantly with rifabutin or rifampin. In vivo data suggest that rifabutin is metabolized in part by CYP3A4. Itraconazole may inhibit the metabolism of rifabutin. Itraconazole may inhibit the metabolism of busulfan, docetaxel and vinca alkaloids. For more Interactions (Complete) data for Itraconazole (29 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Rat oral >320 mg/kg LD50 Mouse oral >320 mg/kg LD50 Dog oral >200 mg/kg LD50 Guinea pig oral >160 mg/kg |
| 参考文献 |
|
| 其他信息 |
Itraconazole is an antifungal prescription medicine approved by the U.S. Food and Drug Administration (FDA) for the treatment of certain fungal infections, such as:
Histoplasmosis Certain types of mucocutaneouscandidiasis, including esophageal candidiasis (infection of the esophagus) and oropharyngeal candidiasis (infection of part of the throat) Histoplasmosis and mucocutaneous candidiasis can be opportunistic infections (OIs) of HIV. First synthesized in the early 1980s, itraconazole is a broad-spectrum triazole antifungal agent used to treat a variety of infections. It is a 1:1:1:1 racemic mixture of four diastereomers, made up of two enantiomeric pairs, each possessing three chiral centers. Itraconazole was first approved in the US in 1992 and is available orally. While the intravenous formulation of the drug was formerly available, it was discontinued in the US in 2007. Itraconazole is a orally administered, triazole antifungal agent used in the treatment of systemic and superficial fungal infections. Itraconazole therapy is associated with transient, mild-to-moderate serum elevations and can lead to clinically apparent acute drug induced liver injury. Itraconazole is a synthetic triazole agent with antimycotic properties. Formulated for both topical and systemic use, itraconazole preferentially inhibits fungal cytochrome P450 enzymes, resulting in a decrease in fungal ergosterol synthesis. Because of its low toxicity profile, this agent can be used for long-term maintenance treatment of chronic fungal infections. (NCI04) A triazole antifungal agent that inhibits cytochrome P-450-dependent enzymes required for ERGOSTEROL synthesis. Drug Indication Itraconazole is indicated for the treatment of the following fungal infections in immunocompromised and non-immunocompromised patients: - Blastomycosis, pulmonary and extrapulmonary - Histoplasmosis, including chronic cavitary pulmonary disease and disseminated, nonmeningeal histoplasmosis, and - Aspergillosis, pulmonary and extrapulmonary, in patients who are intolerant of or who are refractory to amphotericin B therapy It is also indicated for the treatment of the following fungal infections in non-immunocompromised patients: - Onychomycosis of the toenail, with or without fingernail involvement, due to dermatophytes (tinea unguium) - Onychomycosis of the fingernail due to dermatophytes (tinea unguium). Itraconazole oral solution is indicated for the treatment of oropharyngeal and esophageal candidiasis. For the treatment of aspergillosis and candidiasis in companion birds, Mechanism of Action Itraconazole mediates its antifungal activity by inhibiting 14α-demethylase, a fungal cytochrome P450 enzyme that converts lanosterol to ergosterol, a vital component of fungal cell membranes. The azole nitrogen atoms in the chemical structure of itraconazole form a complex with the active site, or the haem iron, of the fungal enzyme to impede its function. The accumulation of lanosterol and 14-methylated sterols results in increased permeability of the fungal cell membrane, and modified membrane-bound enzyme activity, and dysregulated chitin synthesis. Other proposed mechanisms of action of itraconazole include the inhibition of fungal cytochrome c oxidative and peroxidative enzymes that also lead to the disruption of fungal cell membranes. In vitro studies have demonstrated that itraconazole inhibits the cytochrome P450-dependent synthesis of ergosterol, which is a vital component of fungal cell membranes. Therapeutic Uses Antifungal Agents; Antiprotozoal Agents Itraconazole capsules are indicated for the treatment of the following fungal infections in immunocompromised and non-immunocompromised patients: Blastomycosis, pulmonary and extrapulmonary; Histoplasmosis, including chronic cavitary pulmonary disease and disseminated, non-meningeal histoplasmosis and Aspergillosis, pulmonary and extrapulmonary, in patients who are intolerant of or who are refractory to amphotericin B therapy. /Included in US product label/ Itraconazole capsules are also indicated for the treatment of the following fungal infections in non-immunocompromised patients: Onychomycosis of the toenail, with or without fingernail involvement, due to dermatophytes (tinea unguium) and Onychomycosis of the fingernail due to dermatophytes (tinea unguium). /Included in US product label/ Drug Warnings /BOXED WARNING/ Congestive Heart Failure, Cardiac Effects: Itraconazole capsules should not be administered for the treatment of onychomycosis in patients with evidence of ventricular dysfunction such as congestive heart failure (CHF) or a history of CHF. If signs or symptoms of congestive heart failure occur during administration of itraconazole capsules, discontinue administration. When itraconazole was administered intravenously to dogs and healthy human volunteers, negative inotropic effects were seen. /BOXED WARNING/ Drug Interactions: Coadministration of the following drugs are contraindicated with itraconazole capsules: methadone, disopyramide, dofetilide, dronedarone, quinidine, ergot alkaloids (such as dihydroergotamine, ergometrine (ergonovine), ergotamine, methylergometrine (methylergonovine)), irinotecan, lurasidone, oral midazolam, pimozide, triazolam, felodipine, nisoldipine, ranolazine, eplerenone, cisapride, lovastatin, simvastatin and, in subjects with renal or hepatic impairment, colchicine. Coadministration with itraconazole can cause elevated plasma concentrations of these drugs and may increase or prolong both the pharmacologic effects and/or adverse reactions to these drugs. For example, increased plasma concentrations of some of these drugs can lead to QT prolongation and ventricular tachyarrhythmias including occurrences of torsades de pointes, a potentially fatal arrhythmia. Itraconazole is contraindicated in patients with known hypersensitivity to the drug or any ingredient in the formulation. Although information concerning cross-sensitivity between itraconazole and other triazole or imidazole antifungal agents is not available, the manufacturer states that itraconazole should be used with caution in individuals hypersensitive to other azoles. Adverse GI effects have been reported in about 1-11% of patients receiving IV or oral itraconazole for the treatment of systemic fungal infections or oropharyngeal or esophageal candidiasis or for empiric anti-fungal therapy. These adverse GI effects usually are transient and respond to symptomatic treatment without alteration of itraconazole therapy; however, reduction of dosage or discontinuance of the drug occasionally may be required. For more Drug Warnings (Complete) data for Itraconazole (27 total), please visit the HSDB record page. Pharmacodynamics Itraconazole is an antifungal agent that inhibits cell growth and promotes cell death of fungi. It exhibits in vitro activity against _Blastomyces dermatitidis_, _Histoplasma capsulatum_, _Histoplasma duboisii_, _Aspergillus flavus_, _Aspergillus fumigatus_, and _Trichophyton_ species. |
| 分子式 |
C35H33D5CL2N8O4
|
|---|---|
| 分子量 |
710.66
|
| 精确质量 |
709.271
|
| CAS号 |
1217510-38-7
|
| 相关CAS号 |
Itraconazole;84625-61-6
|
| PubChem CID |
45039617
|
| 外观&性状 |
White to light yellow solid powder
|
| 熔点 |
168-170
166.2 °C |
| LogP |
5.707
|
| tPSA |
104.7
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
11
|
| 重原子数目 |
49
|
| 分子复杂度/Complexity |
1120
|
| 定义原子立体中心数目 |
2
|
| SMILES |
[2H]C([2H])([2H])C([2H])([2H])C(C)N1C(=O)N(C=N1)C2=CC=C(C=C2)N3CCN(CC3)C4=CC=C(C=C4)OC[C@H]5CO[C@](O5)(CN6C=NC=N6)C7=C(C=C(C=C7)Cl)Cl
|
| InChi Key |
VHVPQPYKVGDNFY-VXTATNQMSA-N
|
| InChi Code |
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(24-40-45)29-7-5-27(6-8-29)41-14-16-42(17-15-41)28-9-11-30(12-10-28)47-19-31-20-48-35(49-31,21-43-23-38-22-39-43)32-13-4-26(36)18-33(32)37/h4-13,18,22-25,31H,3,14-17,19-21H2,1-2H3/t25?,31-,35-/m0/s1/i1D3,3D2
|
| 化学名 |
4-[4-[4-[4-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-2-(3,3,4,4,4-pentadeuteriobutan-2-yl)-1,2,4-triazol-3-one
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4071 mL | 7.0357 mL | 14.0714 mL | |
| 5 mM | 0.2814 mL | 1.4071 mL | 2.8143 mL | |
| 10 mM | 0.1407 mL | 0.7036 mL | 1.4071 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
A Study to Evaluate the Drug-drug Interaction Effect of Itraconazole on the Pharmacokinetics (PK) of AMG 510 in Healthy Participants
CTID: NCT05568082
Phase: Phase 1 Status: Completed
Date: 2024-11-21
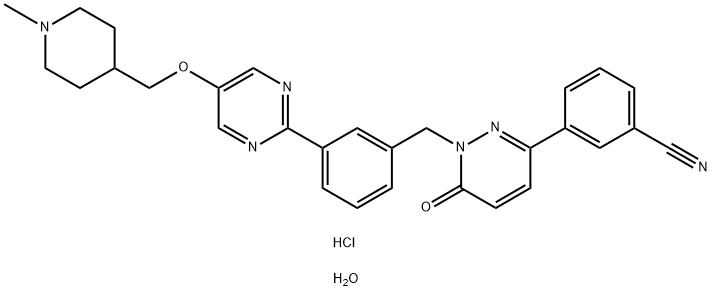 Tepotinib hydrochloride monohydrate
Tepotinib hydrochloride monohydrate
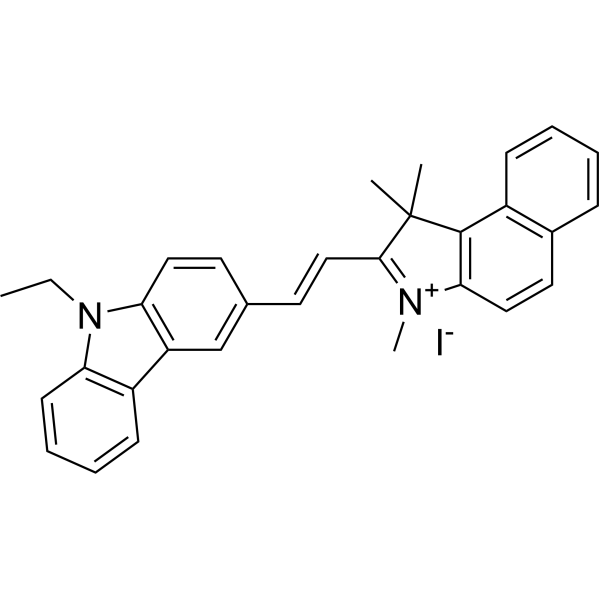 BKN-1
BKN-1
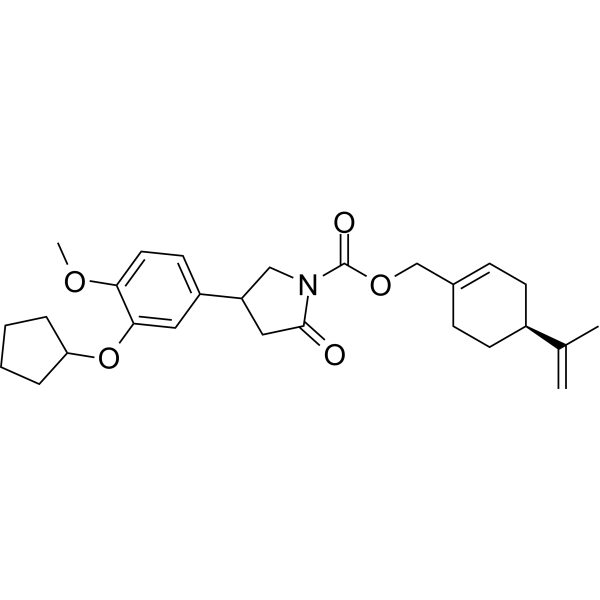 NEO214
NEO214
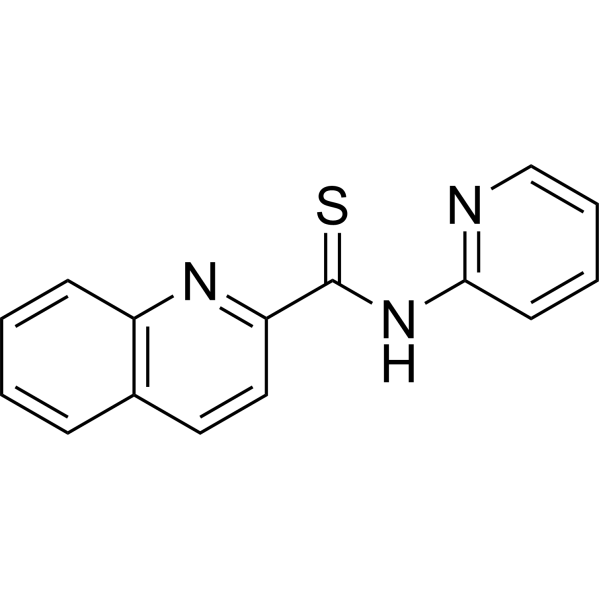 MJO445
MJO445
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























