
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
human COX-1 (IC50 = 18 nM in CHO cells); hCOX-2 (IC50 = 26 nM in CHO cells)
|
|---|---|
| 体外研究 (In Vitro) |
吲哚美辛钠水合物(0-150 μM;24 小时;3LL-D122 细胞)具有体外抗肿瘤活性 [2]。通过激活 PKR 和磷酸化 eLF2α,吲哚美辛钠水合物 (0-1000 μM) 抑制病毒复制 (IC50=2 μM) 并停止病毒蛋白翻译,保护宿主细胞免受病毒伤害 [3]。吲哚美辛氯化钠水合物
|
| 体内研究 (In Vivo) |
卡拉胶诱导的痛觉过敏可被吲哚美辛(吲哚美辛)钠水合物(0.01-10 mg/kg;口服;3 小时;雄性 Sprague-Dawley 大鼠)剂量依赖性地逆转,这也会引起爪子水肿和痛觉过敏 [1]。吲哚美辛(10 mg/mL;口服;每天一次,持续 29 天;雄性 C57BL/6J 小鼠)在体内抑制肿瘤生长 [2]。
使用吲哚美辛(IND),动物胃溃疡模型可生成如下: 胃溃疡模型的生成:所有动物在给药前24 h禁食。除对照组外,IND、IND+ ESP和IND+ CA三个实验研究组均采用IND诱导溃疡。实验动物给予与对照组相同体积的生理盐水。IND给药后6 h给药氯胺酮50 mg/kg,噻嗪5 mg/kg。麻醉后的大鼠颈椎脱臼安乐死,取组织标本。具体地说,胃沿着大弯曲打开,用生理盐水在4°C下清洗。洗净的胃组织保存在含有10%福尔马林的管中进行组织学检查,并在- 800°C下进行生化测定直至分析。用组织病理学和免疫组织化学方法评估所取组织的苏木精-伊红染色。[4] |
| 酶活实验 |
测定Ki和k2值对COX-2的时间依赖性抑制作用[1]
纯化的COX-2 (2.3 μg)与抑制剂在180 μl的反应缓冲液中预孵育0-15 min,然后用花生四烯酸和TMPD的混合物开始反应。用上述分光光度法测定环加氧酶活性。在没有抑制剂预孵育的情况下进行的实验中,将含有酶的测定混合物加入抑制剂和花生四烯酸/TMPD乙醇溶液中引发反应。通过使用Sigmaplot软件将数据拟合到形式为y=a + b.exp(-kobst)的一阶方程中,计算出每种抑制剂浓度下随时间变化的活性损失的速率常数(kobs)。根据Rome and Lands(1975)开发的模型分析了绵羊COX-1的时间依赖性抑制。在该模型(方案1)中,酶和抑制剂的初始可逆结合(以解离常数Ki为特征)之后是一级失活过程(以一级速率常数k2为特征)。该过程的逆转速率(k-2)可以忽略不计。 抑制剂结合的化学计量学测定[1] 等分纯化的COX-2 (0.25 mg ml-1,亚基浓度为3.4 μm)在不同浓度的抑制剂(0-8 μm)存在的缓冲液(100 mm Tris-HCl, pH 8.0, 5 mm EDTA, 1 mm苯酚)中孵育15或30分钟。然后去除等分(20 μl),通过吸氧测定剩余的环加氧酶活性,如上所述。酶的浓度由酸水解后的氨基酸浓度决定(Percival et al., 1994)。 花生四烯酸对COX-2抑制的时间依赖性竞争[1] 将纯化的COX-2 (3.6 μg)稀释到含有60 mm二乙基二硫代氨基甲酸的预孵化液(0.03 ml, 100 mm Tris-HCl, pH 8.0, 5 mm EDTA, 2 mm苯酚)中,以防止底物氧化(Lands et al., 1974)和10 μm抑制剂,或10 μm抑制剂加5 μm花生四烯酸,或10 μm抑制剂加30 μm花生四烯酸。预孵育0-4分钟后,在30°C条件下用耗氧量测定总酶的酶活性,如上所述。 |
| 细胞实验 |
细胞活力测定[2]
细胞类型: 3LL-D122 细胞(小鼠 LL 癌细胞的高度转移变体) 测试浓度: 0、20、50 、100 和 150μM 孵育时间:24 小时 实验结果:20 mM 时抑制细胞活力,60 mM 时抑制 50%。 细胞周期分析[2] 细胞类型: 3LL-D122 细胞(小鼠 LL 癌细胞的高度转移变体) 测试浓度: > 0、30 和 80μM 孵育时间:24 小时 实验结果:G2/M 期细胞百分比减少, G1期细胞百分比增加。 |
| 动物实验 |
Animal/Disease Models: Male SD (Sprague-Dawley) rats[1]
Doses: 0.01-10 mg/kg Route of Administration: Oral administration; for 3 hrs (hours) Experimental Results: Inhibited the carrageenan- induced rat paw oedema (ED50=2.0 mg/kg) and hyperalgesia (ED50=1.5 mg/kg) in a dose-dependent manner. Animal/Disease Models: Male C57BL/6J mice[2] Doses: 10 mg/mL Route of Administration: Oral administration ; daily, for 29 days Experimental Results: Delayed the onset of tumor growth and the initial growth rate of the footpad tumors. |
| 参考文献 |
[1]. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol. 1997 May;121(1):105-17.
[2]. Comparative effects of indomethacin on cell proliferation and cell cycle progression in tumor cells grown in vitro and in vivo. Biochem Pharmacol. 2001 Mar 1;61(5):565-71. [3]. Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: role of eIF2α kinase PKR. Cell Microbiol. 2015 Sep;17(9):1391-404. [4]. Neurogenic transdifferentiation of human adipose-derived stem cells? A critical protocol reevaluation with special emphasis on cell proliferation and cell cycle alterations. Histochem Cell Biol. 2010;134(5):453-468. |
| 其他信息 |
Indomethacin Sodium is the sodium salt of indomethacin, a methylated indole derivative with anti-inflammatory, analgesic-antipyretic and tocolytic effects. Indomethacin is a non-selective, reversible, and competitive inhibitor of cyclooxygenases 1 and 2, thereby blocking the conversion of arachidonic acid into prostaglandin precursors. Consequently, prostaglandin synthesis is decreased, and prostaglandin-mediated activities are prevented, including pain, inflammation, fever and uterine contraction.
Indometacin is a member of the class of indole-3-acetic acids that is indole-3-acetic acid in which the indole ring is substituted at positions 1, 2 and 5 by p-chlorobenzoyl, methyl, and methoxy groups, respectively. A non-steroidal anti-inflammatory drug, it is used in the treatment of musculoskeletal and joint disorders including osteoarthritis, rheumatoid arthritis, gout, bursitis and tendinitis. It has a role as an EC 1.14.99.1 (prostaglandin-endoperoxide synthase) inhibitor, an analgesic, a gout suppressant, a drug metabolite, a xenobiotic metabolite, a xenobiotic, an environmental contaminant and a non-steroidal anti-inflammatory drug. It is a N-acylindole, a member of monochlorobenzenes, an aromatic ether and a member of indole-3-acetic acids. Indometacin, or indomethacin, is a non-steroidal anti-inflammatory drug (NSAID) with anti-inflammatory, analgesic, and antipyretic properties. NSAIDs consist of agents that are structurally unrelated; the NSAID chemical classification of indometacin is an indole-acetic acid derivative with the chemical name 1- (p-chlorobenzoyl)25-methoxy-2-methylindole-3-acetic acid. The pharmacological effect of indometacin is not fully understood, however, it is thought to be mediated through potent and nonselective inhibition of the enzyme cyclooxygenase (COX), which is the main enzyme responsible for catalyzes the rate-limiting step in prostaglandin and thromboxane biosynthesis via the arachidonic acid (AA) pathway. Indometacin was first discovered in 1963 and it was first approved for use in the U.S. by the Food and Drug Administration in 1965, along with other acetic acid derivatives such as [diclofenac] and [sulindac] that were also developed during the 1960s. Since then, indometacin has been extensively studied in clinical trials as one of the most potent NSAIDs in blocking prostaglandin synthesis and was among the first NSAIDs to be used in the symptomatic treatment of migraine and for headaches that eventually became known as “indomethacin-responsive” headache disorders. Most commonly used in rheumatoid arthritis, ankylosing spondylitis, osteoarthritis, acute shoulder pains, and acute gouty arthritis, indometacin is currently available as oral capsules as well as other methods of administration, including rectal and intravenous formulations. Intravenous indometacin is administered to close a hemodynamically significant patent ductus arteriosus, as indicated by clinical evidence, in premature infants. Ophthalmic indometacin has been studied and used in the symptomatic treatment of postoperative ocular inflammation and pain and/or complications after cataract surgery. Although deemed effective in reducing ocular inflammation in clinical studies, topical NSAIDs were also associated with a potential reduction in corneal sensitivity accompanied by an increased risk of superficial punctate keratitis and subjective symptoms of discomfort, including pain, burning or pricking, or a tingling sensation after instillation into the cul‐de‐sac. Indomethacin is a Nonsteroidal Anti-inflammatory Drug. The mechanism of action of indomethacin is as a Cyclooxygenase Inhibitor. Indomethacin is a potent nonsteroidal antiinflammatory drug (NSAID) typically used for chronic inflammatory arthritis. Indomethacin has been associated with rare cases of idiosyncratic drug induced liver disease. View MoreIndomethacin is a synthetic nonsteroidal indole derivative with anti-inflammatory activity and chemopreventive properties. As a nonsteroidal anti-inflammatory drug (NSAID), indomethacin inhibits the enzyme cyclooxygenase, thereby preventing cyclooxygenase-mediated DNA adduct formation by heterocyclic aromatic amines. This agent also may inhibit the expression of multidrug-resistant protein type 1, resulting in increased efficacies of some antineoplastic agents in treating multi-drug resistant tumors. In addition, indomethacin activates phosphatases that inhibit the migration and proliferation of cancer cells and downregulates survivin, which may result in tumor cell Indomethacin is a non-steroidal antiinflammatory agent (NSAIA) with antiinflammatory, analgesic and antipyretic activity. Its pharmacological effect is thought to be mediated through inhibition of the enzyme cyclooxygenase (COX), the enzyme responsible for catalyzes the rate-limiting step in prostaglandin synthesis via the arachidonic acid pathway. A non-steroidal anti-inflammatory agent (NSAID) that inhibits CYCLOOXYGENASE, which is necessary for the formation of PROSTAGLANDINS and other AUTACOIDS. It also inhibits the motility of POLYMORPHONUCLEAR LEUKOCYTES. Drug Indication Oral indometacin is indicated for symptomatic management of moderate to severe rheumatoid arthritis including acute flares of chronic disease, moderate to severe ankylosing spondylitis, moderate to severe osteoarthritis, acute painful shoulder (bursitis and/or tendinitis) and acute gouty arthritis. Intravenous indometacin is indicated to induce closure of a hemodynamically significant patent ductus arteriosus in premature infants weighing between 500 and 1750 g when after 48 hours usual medical management (e.g., fluid restriction, diuretics, digitalis, respiratory support, etc.) is ineffective. Pharmacodynamics Indometacin is an NSAID with analgesic and antipyretic properties that exerts its pharmacological effects by inhibiting the synthesis of factors involved in pain, fever, and inflammation. Its therapeutic action does not involve pituitary-adrenal stimulation. Indometacin primarily works by suppressing inflammation in rheumatoid arthritis by providing relief of pain as well as reducing fever, swelling, and tenderness. This effectiveness has been demonstrated by a reduction in the extent of joint swelling, the average number of joints displaying symptoms of inflammation, and the severity of morning stiffness. Increased mobility was demonstrated by a decrease in total walking time and by improved functional capability seen as an increase in grip strength. In clinical trials, indometacin was shown to be effective in relieving the pain, reducing the fever, swelling, redness, and tenderness of acute gouty arthritis. Due to its pharmacological actions, the use of indometacin is associated with the risk of serious cardiovascular thrombotic events, including myocardial infarction and stroke, as well as gastrointestinal effects such as bleeding, ulceration, and perforation of the stomach or intestines. In a study of healthy individuals, acute oral and intravenous indometacin therapy resulted in a transiently diminished basal and CO2 stimulated cerebral blood flow; this effect disappeared in one study after one week of oral treatment. The clinical significance of this effect has not been established. Compared to other NSAIDs, it is suggested that indometacin is a more potent vasoconstrictor that is more consistent in decreasing cerebral blood flow and inhibiting CO2 reactivity. There have been studies that show indometacin directly inhibiting neuronal activity to some extent in the trigeminocervical complex after either superior salivatory nucleus or dural stimulation. Absorption Indometacin displays a linear pharmacokinetics profile where the plasma concentrations and area under the curve (AUC) are dose-proportional, whereas half-life (T1/2) and plasma and renal clearance are dose-dependent. Indometacin is readily and rapidly absorbed from the gastrointestinal tract. The bioavailability is virtually 100% following oral administration and about 90% of the dose is absorbed within 4 hours. The bioavailability is about 80-90% following rectal administration. The peak plasma concentrations following a single oral dose were achieved between 0.9 ± 0.4 and 1.5 ± 0.8 hours in a fasting state. Despite large intersubject variation as well using the same preparation, peak plasma concentrations are dose-proportional and averaged 1.54 ± 0.76 μg/mL, 2.65 ± 1.03 μg/mL, and 4.92 ± 1.88 μg/mL following 25 mg, 50 mg, and 75 mg single doses in fasting subjects, respectively. With a typical therapeutic regimen of 25 or 50 mg t.i.d., the steady-state plasma concentrations of indomethacin are an average 1.4 times those following the first dose. Route of Elimination Indometacin is eliminated via renal excretion, metabolism, and biliary excretion. It is also subject to enter the enterohepatic circulation through excretion of its glucuronide metabolites into bile followed by resorption of indometacin after hydrolysis. The extent of involvement in the enterohepatic circulation ranges from 27 to 115%. About 60 percent of an oral dosage is recovered in urine as drug and metabolites (26 percent as indomethacin and its glucuronide), and 33 percent in the feces (1.5 percent as indomethacin). DrugBank Volume of Distribution The volume of distribution ranged from 0.34 to 1.57 L/kg following oral, intravenous, or rectal administration of single and multiple doses of indometacin in healthy individuals. Indometacin is distributed into the synovial fluid and is extensively bound to tissues. It has been detected in human breast milk and placenta. Although indometacin has been shown to cross the blood-brain barrier (BBB), its extensive plasma protein binding allows only the small fraction of free or unbound indometacin to diffuse across the BBB. DrugBank Clearance In a clinical pharmacokinetic study, the plasma clearance of indometacin was reported to range from 1 to 2.5 mL/kg/min following oral administration. Metabolism / Metabolites Indometacin undergoes hepatic metabolism involving glucuronidation, O-desmethylation, and N-deacylation. O-desmethyl-indomethacin, N-deschlorobenzoyl-indomethacin, and O-desmethyl-N-deschlorobenzoyl-indomethacin metabolites and their glucuronides are primarily inactive and have no pharmacological activity. Unconjugated metabolites are also detected in the plasma. Its high bioavailability indicates that indometacin is unlikely to be subject to the first-pass metabolism. Biological Half-Life Indometacin disposition from the plasma is reported to be biphasic, with a half-life of 1 hour during the initial phase and 2.6–11.2 hours during the second phase. Interindividual and intraindividual variations are possible due to the extensive and sporadic nature of the enterohepatic recycling and biliary discharge of the drug. The mean half-life of oral indomethacin is estimated to be about 4.5 hours. The disposition of intravenous indometacin in preterm neonates was shown to vary across premature infants. In neonates older than 7 days, the mean plasma half-life of intravenous indometacin was approximately 20 hours, ranging from 15 hours in infants weighing more than 1000 g and 21 hours in infants weighing less than 1000 g. Mechanism of Action Indometacin is a nonspecific and reversible inhibitor of the cyclo-oxygenase (COX) enzyme or prostaglandin G/H synthase. There are two identified isoforms of COX: COX-1 is universally present in most body tissues and is involved in the synthesis of the prostaglandins and thromboxane A2, while COX-2 is expressed in response to injury or inflammation. Constitutively expressed, the COX-1 enzyme is involved in gastric mucosal protection, platelet, and kidney function by catalyzing the conversion of arachidonic acid to prostaglandin (PG) G2 and PGG2 to PGH2. COX-2 is constitutively expressed and highly inducible by inflammatory stimuli. It is found in the central nervous system, kidneys, uterus, and other organs. COX-2 also catalyzes the conversion of arachidonic acid to PGG2 and PGG2 to PGH2. In the COX-2-mediated pathway, PGH2 is further converted to PGE2 and PGI2 (also known as prostacyclin). PGE2 is involved in mediating inflammation, pain, and fever. Decreasing levels of PGE2 leads to reduced inflammatory reactions. Indometacin is known to inhibit both isoforms of COX, however, with greater selectivity for COX-1, which accounts for its increased adverse gastric effects relative to other NSAIDs. It binds to the enzyme's active site and prevents the interaction between the enzyme and its substrate, arachidonic acid. Indometacin, unlike other NSAIDs, also inhibits phospholipase A2, the enzyme responsible for releasing arachidonic acid from phospholipids. The analgesic, antipyretic and anti-inflammatory effects of indomethacin as well as adverse reactions associated with the drug occur as a result of decreased prostaglandin synthesis. Its antipyretic effects may be due to action on the hypothalamus, resulting in increased peripheral blood flow, vasodilation, and subsequent heat dissipation. The exact mechanism of action of indometacin in inducing closure of a patent ductus arteriosus is not fully understood; however, it is thought to be through inhibition of prostaglandin synthesis. At birth, the ductus arteriosus is normally closed as the tension of the oxygen increases significantly after birth. Patent ductus arteriosus in premature infants is associated with congenital heart malformations where PGE1 mediates an opposite effect to that of oxygen. PGE1 dilates the ductus arteriosus through smooth muscle relaxation and prevents the closure of the ductus arteriosus. By inhibiting the synthesis of prostaglandins, indometacin promotes the closure of ductus arteriosus. Indometacin has been described as possessing anticancer and antiviral properties through activation of protein kinase R (PKR) and downstream phosphorylation of eIF2α, inhibiting protein synthesis. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin and indomethacin (IND) are the most commonly prescribed for inflammation or pain. However, widespread use causes several adverse effects, such as gastric ulcers, upper gastric system bleeding, and erosions. Carnosic acid (CA) is an important natural antioxidant found in rosemary (Rosmarinus essentials) and exhibits a protective effect by suppressing oxidative stress and inflammation. This study aimed to investigate the impact of CA on IND-induced gastric ulceration. Wistar male rats received CA (100 mg/kg) or esomeprazole (ESP) (20 mg/kg, standard drug) by oral gavage for 14 days, after that gastric ulceration was induced by oral administration of 100 mg/kg IND. CA pretreatment attenuated both gross morphological lesions and histopathological alterations. CA strongly reduced IND-induced oxidative stress, verified by a decrease in MDA (p < 0.001) and TOS levels (p < 0.05). Furthermore, an IND-dependent increase in CAT (p < 0.001) and GPx (p < 0.01) activities, as well as a reduction in GSH levels (p < 0.01), were ameliorated by CA pretreatment. CA also attenuated inflammatory damage by suppressing IL-1β (p < 0.01), IL-6 (p < 0.01), and TNFα (p < 0.001) production and increasing Nrf2/HO-1 (p < 0.05) expressions. In conclusion, CA shows a gastroprotective effect by reducing oxidative stress and attenuating inflammation.[4] 1. DFU (5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl)phenyl-2(5H)-furan one) was identified as a novel orally active and highly selective cyclo-oxygenase-2 (COX-2) inhibitor. 2. In CHO cells stably transfected with human COX isozymes, DFU inhibited the arachidonic acid-dependent production of prostaglandin E2 (PGE2) with at least a 1,000 fold selectivity for COX-2 (IC50 = 41 +/- 14 nM) over COX-1 (IC50 > 50 microM). Indomethacin was a potent inhibitor of both COX-1 (IC50 = 18 +/- 3 nM) and COX-2 (IC50 = 26 +/- 6 nM) under the same assay conditions. The large increase in selectivity of DFU over indomethacin was also observed in COX-1 mediated production of thromboxane B2 (TXB2) by Ca2+ ionophore-challenged human platelets (IC50 > 50 microM and 4.1 +/- 1.7 nM, respectively). 3. DFU caused a time-dependent inhibition of purified recombinant human COX-2 with a Ki, value of 140 +/- 68 microM for the initial reversible binding to enzyme and a kappa 2 value of 0.11 +/- 0.06 s-1 for the first order rate constant for formation of a tightly bound enzyme-inhibitor complex. Comparable values of 62 +/- 26 microM and 0.06 +/- 0.01 s-1, respectively, were obtained for indomethacin. The enzyme-inhibitor complex was found to have a 1:1 stoichiometry and to dissociate only very slowly (t1/2 = 1-3 h) with recovery of intact inhibitor and active enzyme. The time-dependent inhibition by DFU was decreased by co-incubation with arachidonic acid under non-turnover conditions, consistent with reversible competitive inhibition at the COX active site. 4. Inhibition of purified recombinant human COX-1 by DFU was very weak and observed only at low concentrations of substrate (IC50 = 63 +/- 5 microM at 0.1 microM arachidonic acid). In contrast to COX-2, inhibition was time-independent and rapidly reversible. These data are consistent with a reversible competitive inhibition of COX-1. 5. DFU inhibited lipopolysaccharide (LPS)-induced PGE2 production (COX-2) in a human whole blood assay with a potency (IC50 = 0.28 +/- 0.04 microM) similar to indomethacin (IC50 = 0.68 +/- 0.17 microM). In contrast, DFU was at least 500 times less potent (IC50 > 97 microM) than indomethacin at inhibiting coagulation-induced TXB2 production (COX-1) (IC50 = 0.19 +/- 0.02 microM). 6. In a sensitive assay with U937 cell microsomes at a low arachidonic acid concentration (0.1 microM), DFU inhibited COX-1 with an IC50 value of 13 +/- 2 microM as compared to 20 +/- 1 nM for indomethacin. CGP 28238, etodolac and SC-58125 were about 10 times more potent inhibitors of COX-1 than DFU. The order of potency of various inhibitors was diclofenac > indomethacin approximately naproxen > nimesulide approximately meloxicam approximately piroxicam > NS-398 approximately SC-57666 > SC-58125 > CGP 28238 approximately etodolac > L-745,337 > DFU. 7. DFU inhibited dose-dependently both the carrageenan-induced rat paw oedema (ED50 of 1.1 mg kg-1 vs 2.0 mg kg-1 for indomethacin) and hyperalgesia (ED50 of 0.95 mg kg-1 vs 1.5 mg kg-1 for indomethacin). The compound was also effective at reversing LPS-induced pyrexia in rats (ED50 = 0.76 mg kg-1 vs 1.1 mg kg-1 for indomethacin). 8. In a sensitive model in which 51Cr faecal excretion was used to assess the integrity of the gastrointestinal tract in rats, no significant effect was detected after oral administration of DFU (100 mg kg-1, b.i.d.) for 5 days, whereas chromium leakage was observed with lower doses of diclofenac (3 mg kg-1), meloxicam (3 mg kg-1) or etodolac (10-30 mg kg-1). A 5 day administration of DFU in squirrel monkeys (100 mg kg-1) did not affect chromium leakage in contrast to diclofenac (1 mg kg-1) or naproxen (5 mg kg-1). 9. The results indicate that COX-1 inhibitory effects can be detected for all selective COX-2 inhibitors tested by use of a sensitive assay at low substrate concentration. The novel inhibitor DFU shows the lowest inhibitory potency against COX-1, a consistent high selectivity of inhibition of COX-2 over COX-1 (>300 fold) with enzyme, whole cell and whole blood assays, with no detectable loss of integrity of the gastrointestinal tract at doses >200 fold higher than efficacious doses in models of inflammation, pyresis and hyperalgesia. These results provide further evidence that prostanoids derived from COX-1 activity are not important in acute inflammatory responses and that a high therapeutic index of anti-inflammatory effect to gastropathy can be achieved with a selective COX-2 inhibitor.[1] Considerable research effort is currently being directed towards understanding the mechanisms mediating the antiproliferative effects of non-steroidal anti-inflammatory drugs (NSAIDs) and, more recently, of cyclooxygenase (COX)-2 inhibitors as well. A key question is whether NSAIDs (excluding sulindac) exert their anticarcinogenic effects in vivo by a mechanism that is dependent on their capacity to inhibit COX activity. Some studies with cultured tumor cells in vitro have argued against such a linkage, showing that NSAIDs inhibit cell replication and/or augment apoptosis only at concentrations that exceed those required to inhibit COX activities 10- to 100-fold. The significance of these results for the observed anticarcinogenic effects of NSAIDs in vivo has not yet been evaluated. We addressed this question by comparing, for the same tumor cells, the effects of the NSAID indomethacin on cell growth parameters when the cells were grown in culture to the effects seen in the in vivo growing tumor in the mouse. Indomethacin added to cultured Lewis lung carcinoma cells exerted a potent antiproliferative effect ((3)H thymidine assay) and reduced cell viability (MTT[3-(4,5-dimethyl(thiazol-2-yl)-2,5 diphenyl tetrazolium bromide] assay) at low doses (10-20 microM) in parallel with its inhibitory effect on cellular cyclooxygenase. These effects of indomethacin appeared to arise from a clear antiproliferative shift in the profile of the cell cycle parameters towards a reduced percentage of cells at the S and G(2)/M phases, together with an increased percentage of cells at the G(1) phase. Significantly, similar results were seen when indomethacin was given in vivo at the low dose of 2 mg per kg/day, which blocked blood platelet COX activity and at the same time produced a delay in tumor growth initiation and attenuation of apparent primary tumor growth as well as growth of lung metastases. These results thus provide strong support for the notion that COX inhibition is a major determinant in the antitumorigenic effect of indomethacin in vivo.[2] Indomethacin, a cyclooxygenase-1 and -2 inhibitor widely used in the clinic for its potent anti-inflammatory/analgesic properties, possesses antiviral activity against several viral pathogens; however, the mechanism of antiviral action remains elusive. We have recently shown that indomethacin activates the double-stranded RNA (dsRNA)-dependent protein kinase R (PKR) in human colon cancer cells. Because of the important role of PKR in the cellular defence response against viral infection, herein we investigated the effect of indomethacin on PKR activity during infection with the prototype rhabdovirus vesicular stomatitis virus. Indomethacin was found to activate PKR in an interferon- and dsRNA-independent manner, causing rapid (< 5 min) phosphorylation of eukaryotic initiation factor-2 α-subunit (eIF2α). These events resulted in shutting off viral protein translation and blocking viral replication (IC50 = 2 μM) while protecting host cells from virus-induced damage. Indomethacin did not affect eIF2α kinases PKR-like endoplasmic reticulum-resident protein kinase (PERK) and general control non-derepressible-2 (GCN2) kinase, and was unable to trigger eIF2α phosphorylation in the presence of PKR inhibitor 2-aminopurine. In addition, small-interfering RNA-mediated PKR gene silencing dampened the antiviral effect in indomethacin-treated cells. The results identify PKR as a critical target for the antiviral activity of indomethacin and indicate that eIF2α phosphorylation could be a key element in the broad spectrum antiviral activity of the drug.[3] |
| 分子式 |
C19H21CLNNAO7
|
|---|---|
| 分子量 |
433.82
|
| 精确质量 |
357.076
|
| 元素分析 |
C, 52.60; H, 4.88; Cl, 8.17; N, 3.23; Na, 5.30; O, 25.82
|
| CAS号 |
74252-25-8
|
| 相关CAS号 |
Indomethacin;53-86-1;Indomethacin-d4;87377-08-0;Indomethacin;53-86-1;Indomethacin sodium;7681-54-1; Indomethacin sodium hydrate;74252-25-8; 7681-54-1 (sodium)
|
| PubChem CID |
23674731
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
499.4±45.0 °C at 760 mmHg
|
| 熔点 |
162ºC
|
| 闪点 |
255.8±28.7 °C
|
| 蒸汽压 |
0.0±1.3 mmHg at 25°C
|
| 折射率 |
1.619
|
| LogP |
3.11
|
| tPSA |
99.05
|
| InChi Key |
UHYAQBLOGVNWNT-UHFFFAOYSA-M
|
| InChi Code |
InChI=1S/C19H16ClNO4.Na.3H2O/c1-11-15(10-18(22)23)16-9-14(25-2)7-8-17(16)21(11)19(24)12-3-5-13(20)6-4-12;;;;/h3-9H,10H2,1-2H3,(H,22,23);;3*1H2/q;+1;;;/p-1
|
| 化学名 |
sodium;2-[1-(4-chlorobenzoyl)-5-methoxy-2-methylindol-3-yl]acetate;trihydrate
|
| 别名 |
Indomethacin sodium trihydrate; 74252-25-8; Indomethacin sodium salt trihydrate; Indometacin sodium; Indomethacin sodium hydrate; 0IMX38M2GG; Indometacin (sodium hydrate); Indometacin sodium trihydrate;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: (1). This product requires protection from light (avoid light exposure) during transportation and storage. (2). Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O : 25 mg/mL (57.63 mM)
DMSO : 12.5 mg/mL (28.81 mM ) |
|---|---|
| 溶解度 (体外实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.79 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.79 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: 5 mg/mL (11.53 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶 (<60°C). 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3051 mL | 11.5255 mL | 23.0510 mL | |
| 5 mM | 0.4610 mL | 2.3051 mL | 4.6102 mL | |
| 10 mM | 0.2305 mL | 1.1526 mL | 2.3051 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
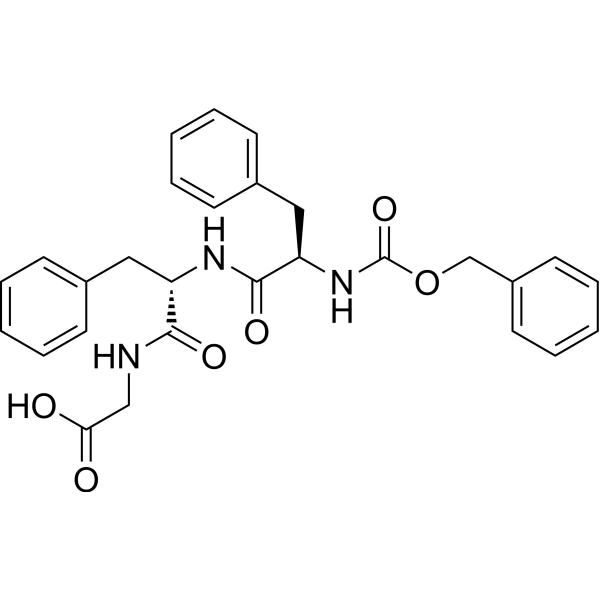 Fusion Inhibitory Peptide (ZD-Phe-Phe-Gly-OH; FIP; Virus Replication Inhibitory Peptide)
Fusion Inhibitory Peptide (ZD-Phe-Phe-Gly-OH; FIP; Virus Replication Inhibitory Peptide)
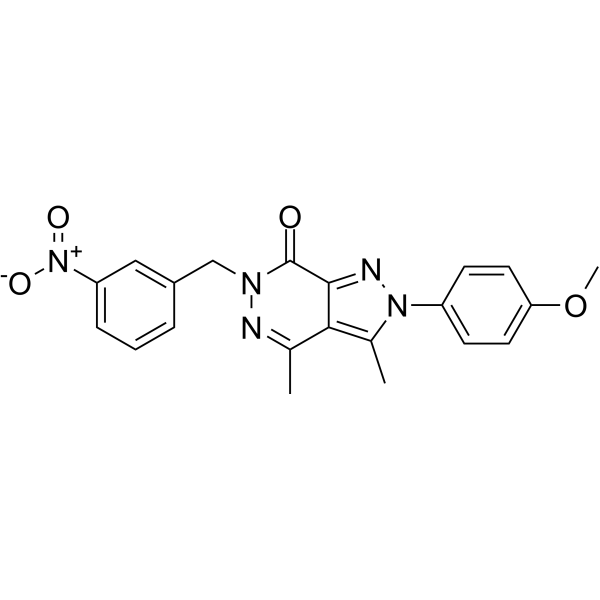 ZIKV-IN-8
ZIKV-IN-8
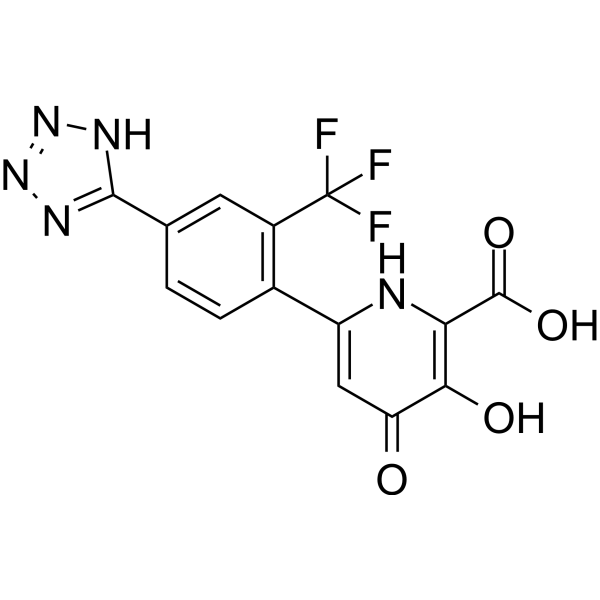 PAN endonuclease-IN-1
PAN endonuclease-IN-1
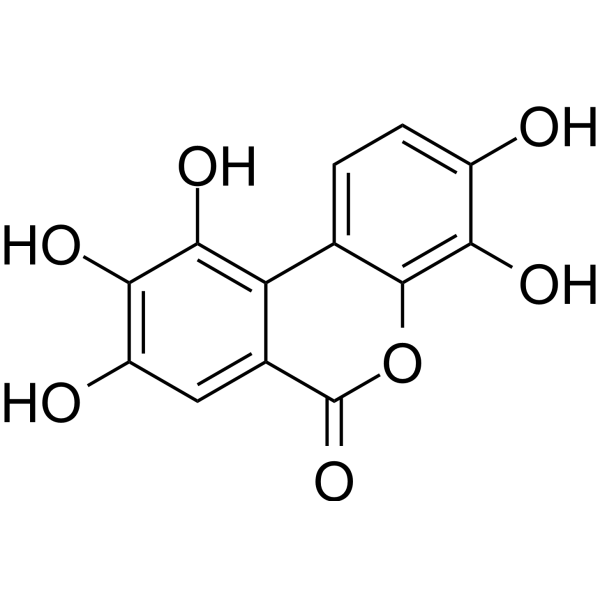 Urolithin M5 (Decarboxyellagic acid)
Urolithin M5 (Decarboxyellagic acid)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
























