
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
FAP (fibroblast activation protein)
|
|---|---|
| 体外研究 (In Vitro) |
在产生人 FAP 的 HT-1080 细胞中,FAPI-46(1-24 小时)短暂地与人 FAP 结合 [1]。
|
| 体内研究 (In Vivo) |
FAPI-46 (iv) 极大地改善了肝脏、肠道和肠道中肿瘤与食物的比例,从而促进了 PET 成像中的图像恢复 [1]。
|
| 细胞实验 |
细胞培养[1]
转染人FAP基因的HT-1080细胞,以及小鼠FAP和cd26转染的人胚胎肾细胞,在含有10%胎牛血清的Dulbecco修饰Eagle培养基中,在37°C/5%二氧化碳条件下培养。 对于放射性配体结合研究,将细胞接种于6孔板中,培养48 h,最终汇合率约为80%-90%(每孔120 - 20万个细胞)。用1 mL不含胎牛血清的新鲜培养基代替培养基。放射性标记的化合物添加到细胞培养,培养不同的间隔10分钟至24小时。竞争实验由同时暴露在无标号(10−5到10−10米)和放射性标记的化合物60分钟。细胞流出决心孵化后的细胞示踪60分钟。此后,放射性介质被移除,细胞被洗和孵化与非放射性介质1,2,4,24 h。在所有的实验中,细胞用1ml pH为7.4的磷酸盐缓冲盐水洗涤两次,然后用1.4 mL裂解缓冲液(0.3 m NaOH, 0.2%十二烷基硫酸钠)裂解。在γ-计数器中测定放射性,归一化到100万个细胞,并以施加剂量的百分比计算。每个实验进行3次,每个独立实验进行3次重复。 |
| 动物实验 |
Animal Studies[1]
For in vivo experiments, 8-wk-old BALB/c nu/nu mice were subcutaneously inoculated into the right trunk with 5 million HT-1080-FAP cells. When the size of the tumor reached approximately 1 cm3, the radiolabeled compound was injected via the tail vein (80 nmol/GBq for small-animal PET imaging; 200 nmol/GBq for organ distribution). In vivo blocking experiments were performed by adding 30 nmol of unlabeled FAPI to the radiolabeled compound directly before injection. For organ distribution, the animals (n = 3 for each time point) were killed 1, 4, 6, and 24 h after tracer administration. The distributed radioactivity was measured in all dissected organs and in blood using a γ-counter. The values are expressed as percentage injected dose per gram of tissue (%ID/g). PET imaging was performed using a small-animal PET scanner. Within the first 60 min, a dynamic scan was performed in list mode, followed by a static scan from 120 to 140 min after injection. Images were reconstructed iteratively using the 3-dimensional ordered-subset expectation maximization maximum a priori method and were converted to SUV images. For the dynamic data, 28 frames were reconstructed: 4 × 5 s, 4 × 10 s, 4 × 20 s, 4 × 60 s, 4 × 120 s, 6 × 300 s, and 2 × 470 s. Quantification was done using a region-of-interest technique and expressed as SUV. All animal experiments were conducted in compliance with the German animal protection laws (approval 35-91185.81/G-158/15).[1] Clinical PET/CT Imaging[1] Imaging of 8 patients was performed under the conditions of the updated Declaration of Helsinki, section 37 (unproven interventions in clinical practice) and in accordance with the German Pharmaceuticals Law, section 13 (2b), for medical reasons using 68Ga-FAPI-21 and -46, which were applied intravenously (20 nmol, 210–267 MBq for FAPI-21 and 216–242 MBq for FAPI-46). Imaging took place at 10 min, 1 h, and 3 h after tracer administration. The PET/CT scans were obtained with a Biograph mCT Flow PET/CT scanner using the following parameters: slice thickness of 5 mm, increment of 3–4 mm, soft-tissue reconstruction kernel, and CARE Dose. Immediately after CT scanning, a whole-body PET scan was acquired in 3 dimensions (matrix, 200 × 200) in FlowMotion with 0.7 cm/min. The emission data were corrected for randoms, scatter, and decay. Reconstruction was conducted with an ordered-subset expectation maximization algorithm with 2 iterations and 21 subsets and Gauss-filtered to a transaxial resolution of 5 mm in full width at half maximum. Attenuation was corrected using the low-dose nonenhanced CT data. SUVs were quantitatively assessed using a region-of-interest technique. The data were analyzed retrospectively with approval of the local ethics committee (approval S016/2018). |
| 药代性质 (ADME/PK) |
Serum Stability[1]
Processed and solvent-free radioactive compounds (177Lu-FAPI-21 and 177Lu-FAPI-46) were incubated in human sera at 37°C. After the respective incubation time, samples were taken, freed from proteins by precipitation with acetonitrile, and centrifuged, and the supernatant was analyzed via radio–high-performance liquid chromatography. Supplemental Figure 1 shows that even at 24 h, only the initial (radioactive) peaks were detected and neither radioactive degradation products nor free radioactivity were observed. These findings demonstrate that both substances were unhampered by enzymatic components of human sera. |
| 参考文献 |
|
| 其他信息 |
Cancer-associated fibroblasts constitute a vital subpopulation of the tumor stroma and are present in more than 90% of epithelial carcinomas. The overexpression of the serine protease fibroblast activation protein (FAP) allows a selective targeting of a variety of tumors by inhibitor-based radiopharmaceuticals (FAPIs). Of these compounds, FAPI-04 has been recently introduced as a theranostic radiotracer and demonstrated high uptake into different FAP-positive tumors in cancer patients. To enable the delivery of higher doses, thereby improving the outcome of a therapeutic application, several FAPI variants were designed to further increase tumor uptake and retention of these tracers. Methods: Novel quinoline-based radiotracers were synthesized by organic chemistry and evaluated in radioligand binding assays using FAP-expressing HT-1080 cells. Depending on their in vitro performance, small-animal PET imaging and biodistribution studies were performed on HT-1080-FAP tumor-bearing mice. The most promising compounds were used for clinical PET imaging in 8 cancer patients. Results: Compared with FAPI-04, 11 of 15 FAPI derivatives showed improved FAP binding in vitro. Of these, 7 compounds demonstrated increased tumor uptake in tumor-bearing mice. Moreover, tumor-to-normal-organ ratios were improved for most of the compounds, resulting in images with higher contrast. Notably two of the radiotracers, FAPI-21 and -46, displayed substantially improved ratios of tumor to blood, liver, muscle, and intestinal uptake. A first diagnostic application in cancer patients revealed high intratumoral uptake of both radiotracers already 10 min after administration but a higher uptake in oral mucosa, salivary glands, and thyroid for FAPI-21. Conclusion: Chemical modification of the FAPI framework enabled enhanced FAP binding and improved pharmacokinetics in most of the derivatives, resulting in high-contrast images. Moreover, higher doses of radioactivity can be delivered while minimizing damage to healthy tissue, which may improve therapeutic outcome.[1]
Background: Fibroblast activation protein (FAP) is a proline selective serine protease that is overexpressed in tumor stroma and in lesions of many other diseases that are characterized by tissue remodeling. In 2014, a most potent FAP-inhibitor (referred to as UAMC1110) with low nanomolar FAP-affinity and high selectivity toward related enzymes such as prolyl oligopeptidase (PREP) and the dipeptidyl-peptidases (DPPs): DPP4, DPP8/9 and DPP2 were developed. This inhibitor has been adopted recently by other groups to create radiopharmaceuticals by coupling bifunctional chelator-linker systems. Here, we report squaric acid (SA) containing bifunctional DATA5m and DOTA chelators based on UAMC1110 as pharmacophor. The novel radiopharmaceuticals DOTA.SA.FAPi and DATA5m.SA.FAPi with their non-radioactive derivatives were characterized for in vitro inhibitory efficiency to FAP and PREP, respectively and radiochemical investigated with gallium-68. Further, first proof-of-concept in vivo animal study followed by ex vivo biodistribution were determined with [68Ga]Ga-DOTA.SA.FAPi. Results: [68Ga]Ga-DOTA.SA.FAPi and [68Ga]Ga-DATA5m.SA.FAPi showed high complexation > 97% radiochemical yields after already 10 min and high stability over a period of 2 h. Affinity to FAP of DOTA.SA.FAPi and DATA5m.SA.FAPi and its natGa and natLu-labeled derivatives were excellent resulting in low nanomolar IC50 values of 0.7-1.4 nM. Additionally, all five compounds showed low affinity for the related protease PREP (high IC50 with 1.7-8.7 μM). First proof-of-principle in vivo PET-imaging animal studies of the [68Ga]Ga-DOTA.SA.FAPi precursor in a HT-29 human colorectal cancer xenograft mouse model indicated promising results with high accumulation in tumor (SUVmean of 0.75) and low background signal. Ex vivo biodistribution showed highest uptake in tumor (5.2%ID/g) at 60 min post injection with overall low uptake in healthy tissues. Conclusion: In this work, novel PET radiotracers targeting fibroblast activation protein were synthesized and biochemically investigated. Critical substructures of the novel compounds are a squaramide linker unit derived from the basic motif of squaric acid, DOTA and DATA5m bifunctional chelators and a FAP-targeting moiety. In conclusion, these new FAP-ligands appear promising, both for further research and development as well as for first human application.[2] |
| 分子式 |
C41H57F2N11O9
|
|---|---|
| 分子量 |
885.97
|
| 精确质量 |
885.43
|
| 元素分析 |
C, 55.58; H, 6.49; F, 4.29; N, 17.39; O, 16.25
|
| CAS号 |
2374782-04-2
|
| PubChem CID |
139400499
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.45±0.1 g/cm3(Predicted)
|
| 沸点 |
1148.7±65.0 °C(Predicted)
|
| LogP |
-6.7
|
| tPSA |
238
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
19
|
| 可旋转键数目(RBC) |
16
|
| 重原子数目 |
63
|
| 分子复杂度/Complexity |
1620
|
| 定义原子立体中心数目 |
1
|
| SMILES |
CN(CCCN1CCN(CC1)C(=O)CN2CCN(CCN(CCN(CC2)CC(=O)O)CC(=O)O)CC(=O)O)C3=CC4=C(C=CN=C4C=C3)C(=O)NCC(=O)N5CC(C[C@H]5C#N)(F)F
|
| InChi Key |
SDBGUEFOSXNKBX-HKBQPEDESA-N
|
| InChi Code |
InChI=1S/C41H57F2N11O9/c1-47(30-3-4-34-33(21-30)32(5-6-45-34)40(63)46-24-35(55)54-29-41(42,43)22-31(54)23-44)7-2-8-48-17-19-53(20-18-48)36(56)25-49-9-11-50(26-37(57)58)13-15-52(28-39(61)62)16-14-51(12-10-49)27-38(59)60/h3-6,21,31H,2,7-20,22,24-29H2,1H3,(H,46,63)(H,57,58)(H,59,60)(H,61,62)/t31-/m0/s1
|
| 化学名 |
2-[4,7-bis(carboxymethyl)-10-[2-[4-[3-[[4-[[2-[(2S)-2-cyano-4,4-difluoropyrrolidin-1-yl]-2-oxoethyl]carbamoyl]quinolin-6-yl]-methylamino]propyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetrazacyclododec-1-yl]acetic acid
|
| 别名 |
FAPI-46; 2374782-04-2; 59QC5DY68A; UNII-59QC5DY68A; (10-(2-(4-(3-((4-(((2-((2S)-2-Cyano-4,4-difluoro-1-pyrrolidinyl)-2-oxoethyl)amino)carbonyl)-6-quinolinyl)methylamino)propyl)-1-piperazinyl)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetato(3-)-kappaN1,kappaN4,kappaN7,kappaN10)-; 2-[4,7-bis(carboxymethyl)-10-[2-[4-[3-[[4-[[2-[(2S)-2-cyano-4,4-difluoropyrrolidin-1-yl]-2-oxoethyl]carbamoyl]quinolin-6-yl]-methylamino]propyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetrazacyclododec-1-yl]acetic acid; [10-[2-[4-[3-[[4-[[[2-[(2S)-2-Cyano-4,4-difluoro-1-pyrrolidinyl]-2-oxoethyl]amino]carbonyl]-6-quinolinyl]methylamino]propyl]-1-piperazinyl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetato(3-)-kappaN1,kappaN4,kappaN7,kappaN10]-; SCHEMBL21257093;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O : ~100 mg/mL (~112.87 mM)
DMSO : ~100 mg/mL (~112.87 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 5.75 mg/mL (6.49 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,将 100 μL 57.5 mg/mL 澄清 DMSO 储备液加入 900 μL 20% SBE-β-CD 生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: ≥ 5 mg/mL (5.64 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 50.0 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.1287 mL | 5.6435 mL | 11.2871 mL | |
| 5 mM | 0.2257 mL | 1.1287 mL | 2.2574 mL | |
| 10 mM | 0.1129 mL | 0.5644 mL | 1.1287 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
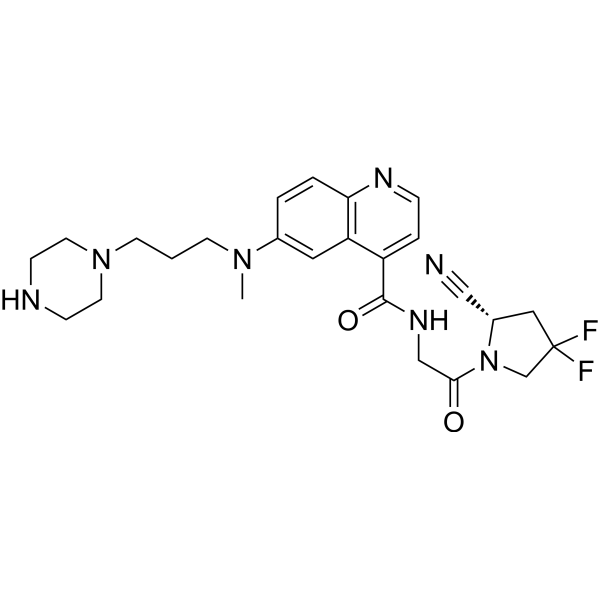 FT-FAPI-12_9
FT-FAPI-12_9
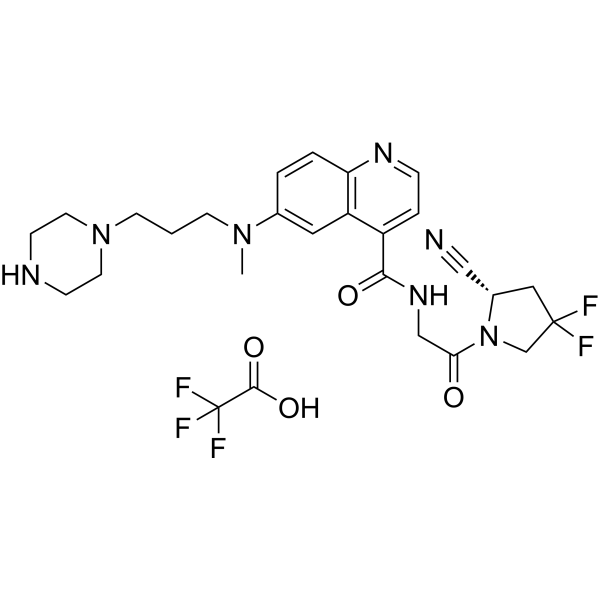 FT-FAPI-12_9 TFA
FT-FAPI-12_9 TFA
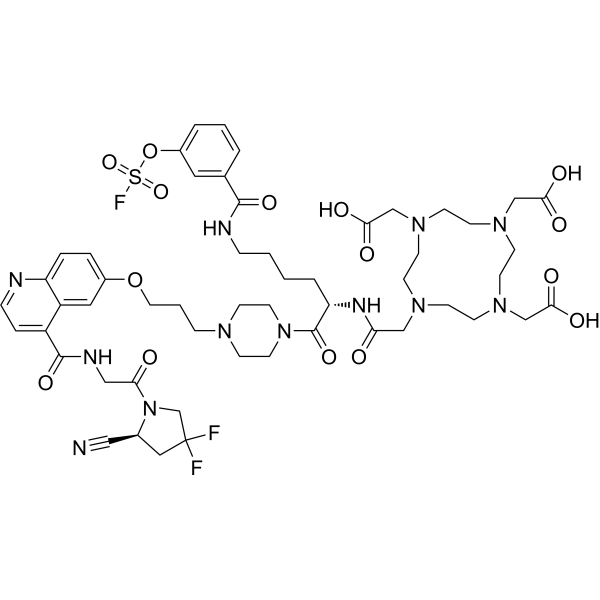 FAPI-mFS
FAPI-mFS
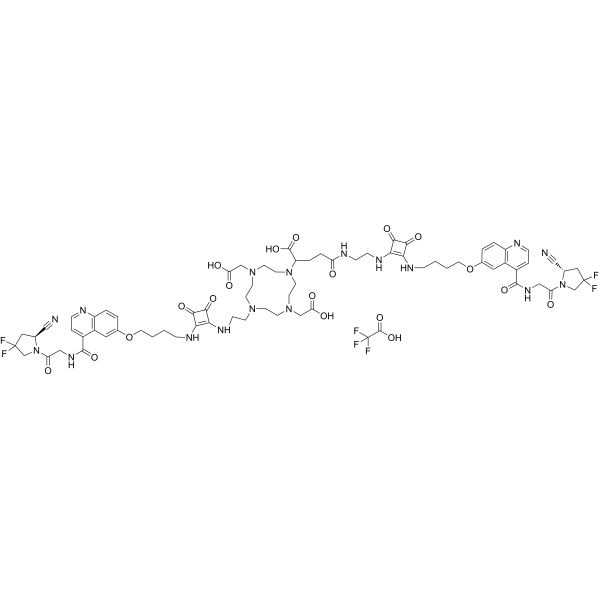 DOTAGA.(SA.FAPi)2 TFA
DOTAGA.(SA.FAPi)2 TFA
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























